Chapter 7 (Updated 6/18/10)
Type 1 Diabetes Mellitus of Man: Genetic
Susceptibility and Resistance
A. K. Steck1, A. Pugliese2 and G.S.
Eisenbarth1
1.
2.
Diabetes Research Institute,
INTRODUCTION
Insulin-dependent diabetes mellitus (IDDM), or type 1
diabetes, is a chronic disease characterized by the autoimmune destruction
(Type 1 A) of pancreatic ß-cells and severe insulin deficiency 1-3. Completion
of multiple large scale genome wide association studies has provided a clearer
understanding of the genetic architecture of Type 1A diabetes4-6. In particular the overwhelming
genetic determinants if Type 1A diabetes are in the major histocompatibility
complex 4, 7. This is followed by insulin
gene polymorphisms, the T cell receptors signaling molecule PTPN22, and the
multiple (>40) loci with very small effects. Of note, there appears to be
little or no overlap between loci for Type 2 and Type 1 diabetes 8Type 1B diabetes refers to insulin dependent diabetes not of immune
etiology, is not the subject of this chapter and has been difficult to
diagnose. It has been suggested that
fulminant diabetes, found almost exclusively in
Type 1A diabetes frequently develops in children,
adolescents and young adults, but approximately half of individuals developing
type 1A diabetes first present as adults. The disease is quite heterogeneous in
its clinical expression and it can be confused with type 2 diabetes, especially
in those patients who develop diabetes at a later age 16, 17. Inherited genetic factors influence both
susceptibility to and resistance to the disease. Although a significant
proportion of patients with type 1A diabetes lack a positive family history for
the disease(>85%), there is significant familial clustering with an average
prevalence of 6% in siblings compared to 0.4% in the US Caucasian population.
The familial clustering (λs) can be calculated as the ratio of the risk to
siblings over the disease prevalence in the general population, and thus
λs = 6/0.4 = 15 18, 19.
One’s
genetic susceptibility depends on the degree of genetic identity with the proband.
The risk of diabetes in family members has a non-linear correlation with the
number of alleles shared with the proband. The highest risk is observed in
monozygotic twins (100% sharing) followed by first, second and third degree
relatives (50%, 25%, 12.5% sharing, respectively). Based on such estimates of
observed risk, it has been suggested that diabetes susceptibility may be linked
to a major locus and that several other minor loci may contribute to diabetes
risk in an epistatic way. This model generates the risk curve that best
parallels the risk curve obtained from observed risk estimates 20. The
moderate disease concordance observed even amongst identical twins (usually
30-50%, 70% in studies with longest follow-up) implies that inherited genes
provide increased susceptibility 21-25.
Much
technological progress has facilitated the study of the genome to map disease
susceptibility genes for multi-factorial diseases, including the increasing
availability of microsatellite markers, single nucleotide polymorphisms (SNPs),
automated typing technology 26, and recently whole genome
SNP analysis 27. In the case
of type 1 diabetes, genome scans for IDDM susceptibility loci have been
facilitated by the availability of large c
It is also
possible that the disease is genetically heterogeneous, with different major
loci determining disease risk in different families. Genetic heterogeneity has
been demonstrated in most of the genome wide scans performed to date. The genetic heterogeneity can also be demonstrated with the
study of groups of monozygotic twins.
When the first twin of a twin-pair develops type 1 diabetes after age
25, the risk of the second monozygotic twin developing type 1 diabetes is less
than 5% with long-term follow up 24, while
approximately 60% of initially discordant twins whose twin mate developed
diabetes prior to age 6 have progressed to diabetes (by life table analysis
with 40 years of follow-up). For
monozygotic twins of patients with type 1 diabetes, expression of anti-islet
autoantibodies directly correlates with progression to overt diabetes. Essentially all such twins who express
“biochemical” anti-islet autoantibodies (to GAD, IA-2/ICA512, insulin, measured
by radioimmunoassays) progress to diabetes even after decades of follow-up 31. In contrast, dizygotic twins have a low risk
of expressing anti-islet autoantibodies, a risk that is essentially identical
to that of siblings. These risk
estimates have been validated through the exchange of sera 32 and confirmed
by a large study of the DPT-1 (Diabetes Prevention Trial – Type 1) cohort of at-risk relatives 24. Similar results were obtained studying a
population-based twin cohort of 22,650 twin pairs from
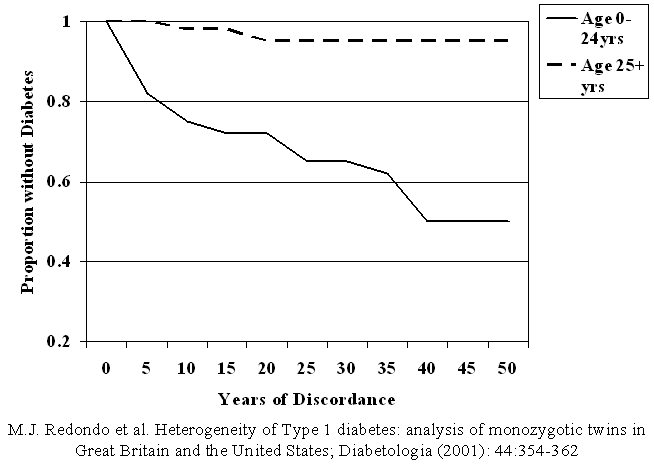
Figure 7.1 Diabetes-free
survival analysis of the combined Great Britain and United States cohorts, by
age at diagnosis in the index twin: Ages 0-24 years (n=150) in solid line, 25
years and older (n=37) in dashed line.
Besides
inherited alleles, other mechanisms regulating gene expression including
epigenetic and parent-of-origin effects influence susceptibility by modifying
the transmission and transcription of inherited genes. It is also an intriguing possibility that
additional genetic factors or their expression may be acquired after birth,
perhaps through environmental exposures.
Thus, a variety of genetic mechanisms may influence the autoimmune
responses leading to ß-cell destruction. This chapter will review the current
knowledge about the genetics of type 1 diabetes in humans.
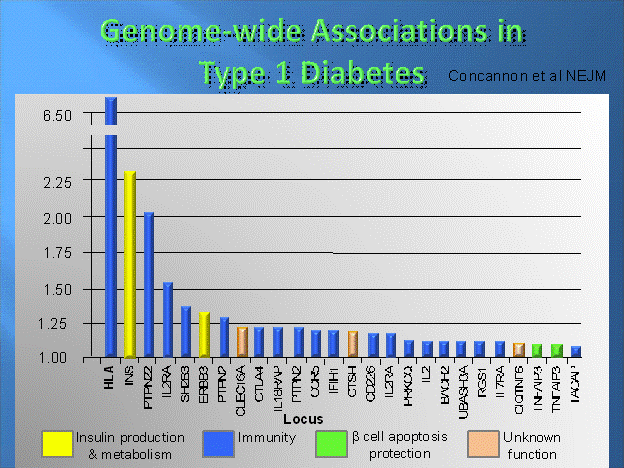
Figure
7.2. Odds ratios for a series of
identified “genes/genetic loci” from recent genome screens and replication
studies.
INHERITED
SUSCEPTIBILITY LOCI
Both association studies and linkage analysis using various
analytical methods have been used to identify IDDM susceptibility loci. These are conventionally noted using the
abbreviation IDDM and a number, e.g. IDDM1,
IDDM2, etc., with the number usually
reflecting the order in which such loci were reported (Table 7.1 and Figure
7.2). Many of the early IDDM loci appear at present to have been “false
positives” and are generally being replaced by identified genes (figure 7.2),
though small influences on diabetes susceptibility that cannot presently be
proven for such loci with the current large genome screens is a
possibility. Using the candidate gene
approach, association studies provided evidence for the first two
susceptibility loci, the HLA region (IDDM1)
and the insulin gene (INS) locus (IDDM2). These two loci contribute the
great majority of known familial clustering (Figure 7.2), suggesting the existence of additional loci. One estimate is
that the MHC alone contributes 41% of the familial clustering of type 1 diabetes
of the 48% estimated to be accounted for with all known genes 29. The next most potent locus for type 1
diabetes of man was also discovered using a candidate gene approach, namely the
PTPN22 (LYP) gene with an odds ratio of approximately 1.7 for a “missense”
mutation that creates susceptibility to multiple autoimmune disorders 34-36. Figure 7.3
illustrates odds ratio for multiple loci recently summarized for GWAS studies. The ratio of differences in frequencies,
except for PTPN22 are relatively small, making it unlikely that the other
indicated loci will contribute to the genetic prediction of type 1A diabetes,
in contrast to the HLA and insulin region genes. For instance the HLA DR3/4-DQ2/8 genotype is
present in 2.3% of newborns in
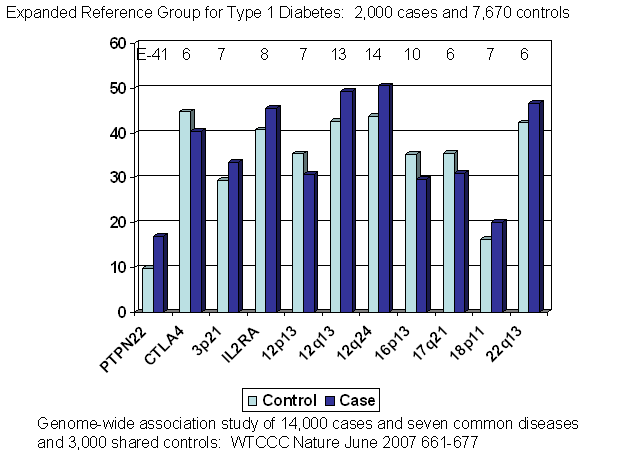
Figure 7.3. Allele
frequencies for case versus control association studies with “significant”
associations outside of the major histocompatibility complex.
Prior to the whole genome SNP analyses that have recently
been reported, a number of genome-wide studies of families and affected
sibling-pairs have been performed since the mid 1990’s in an attempt to
identify susceptibility loci using linkage analysis 44. Linkage analysis confirmed linkage with IDDM1 and IDDM2 and further provided evidence for the existence of
approximately 20 susceptibility loci. Many of these loci show modest linkage
and linkage is often not confirmed in all genome scans. Sample size and
composition, genetic heterogeneity and analytical methods underlie much of the
variability observed in these studies. A coordinated effort to investigate the
genetics of the disease, the Type 1 Diabetes Genetics Consortium (T1DGC) (www.t1dgc.org), has been launched and involves
the study of patients and their families from around the world. In 2005 the
consortium published its first report, an interim, combined
linkage analysis of four datasets, three previously published genome
scans, and a new dataset of 254 families. This analysis included 1,435 families
with 1,636 affected sibling pairs, representing one of the largest
linkage studies ever performed for any common disease and involving families
from the ![]() S
S
![]() 1.3
and p= 10-4. With this analytical power, more than 80% of
the genome was found not to harbor susceptibility genes of modest effect that
could be detected by linkage. The study confirmed linkage with IDDM1
(nominal P = 2.0 x 10–52). Moreover, nine non–HLA-linked
regions showed some evidence of linkage (nominal P <
0.01), including three at (or near) genome-wide significance (P <
0.05): 2q31-q33, 10p14-q11, and 16q22-q24. In addition, after taking
into account the linkage at the 6p21 (HLA) region, there was evidence of linkage
with the 6q21 region (IDDM15). The published literature on these loci is
discussed in detail in the following paragraphs. A comprehensive list of these
initial susceptibility loci is shown in Table 7.1 with LOD scores and
1.3
and p= 10-4. With this analytical power, more than 80% of
the genome was found not to harbor susceptibility genes of modest effect that
could be detected by linkage. The study confirmed linkage with IDDM1
(nominal P = 2.0 x 10–52). Moreover, nine non–HLA-linked
regions showed some evidence of linkage (nominal P <
0.01), including three at (or near) genome-wide significance (P <
0.05): 2q31-q33, 10p14-q11, and 16q22-q24. In addition, after taking
into account the linkage at the 6p21 (HLA) region, there was evidence of linkage
with the 6q21 region (IDDM15). The published literature on these loci is
discussed in detail in the following paragraphs. A comprehensive list of these
initial susceptibility loci is shown in Table 7.1 with LOD scores and ![]() S
from the 2005 T1DGC scan 45.
S
from the 2005 T1DGC scan 45.
Table 7.1. Susceptibility Loci for Type 1 Diabetes as of
“2005”
|
Locus |
Chromosome |
Candidate
Genes |
Markers |
LOD
|
|
|
IDDM1 |
6p21.3 |
HLA DR/DQ
|
TNFA |
116.38 |
3.35 |
|
IDDM2 |
11p15.5 |
INSULIN VNTR
|
D11S922 |
1.87 |
1.16 |
|
PTPN22 |
1p13 |
PTPN22 (LYP)
|
SNP=R620W |
NR |
1.05 |
|
SUMO4 |
6q25 (IDDM5) |
SUMO4
|
SNP=M55VA allele 163 [G] |
NR |
NR |
|
IDDM3 |
15q26 |
|
D15S107 |
NR |
NR |
|
IDDM4 |
11q13.3 |
MDU1, ZFM1, RT6, ICE, LRP5, FADD, CD3 |
FGF3, D11S1917 |
NR |
NR |
|
IDDM5 |
6q25 |
SUMO4,MnSOD |
ESR, a046Xa9 |
NR |
NR |
|
IDDM6 |
18q12-q21 |
JK (Kidd), ZNF236 |
D18S487, D18S64 |
NR |
NR |
|
IDDM7 |
2q31-33 |
NEUROD
|
D2S152, D251391 |
3.34* |
1.19* |
|
IDDM8 |
6q25-27 |
|
D6S281, D6S264, D6S446 |
NR |
NR |
|
IDDM9 |
3q21-25 |
|
D3S1303, D10S193 |
NR |
NR |
|
IDDM10 |
10p11-q11 |
|
D10S1426, D10S565 |
3.21 |
1.12 |
|
IDDM11 |
14q24.3-q31 |
ENSA, SEL-1L |
D14S67 |
NR |
NR |
|
IDDM12 |
2q33 |
CTLA-4 |
(AT)n
3 |
3.34 |
1.19 |
|
IDDM13 |
2q34 |
IGFBP2, IGFBP5, NEUROD, HOXD8 |
D2S137, D2S164, D2S1471 |
NR |
NR |
|
IDDM15 |
6q21 |
|
D6S283, D6S434, D6S1580 |
22.39 |
1.56 |
|
IDDM16 |
14q32.3 |
IGH |
|
NR |
NR |
|
IDDM17 |
10q25 |
|
D10S1750, D10S1773 |
NR |
NR |
|
IDDM18 |
5q31.1-33.1 |
IL-12B |
IL12B |
NR |
NR |
|
|
1q42 |
|
D1S1617 |
NR |
NR |
|
|
16p12-q11.1 |
|
D16S3131 |
1.88 |
1.17 |
|
|
16q22-q24 |
|
D16S504 |
2.64 |
1.19 |
|
|
17q25 |
|
|
NR |
NR |
|
|
19q11 |
|
|
NR |
NR |
|
|
3p13-p14 |
|
D3S1261 |
1.52 |
1.15 |
|
|
9q33-q34 |
|
D9S260 |
2.20 |
1.13 |
|
|
12q14-q12 |
|
D12S375 |
1.66 |
1.10 |
|
|
19p13.3-p.13.2 |
|
INSR |
1.92 |
1.15 |
The recent whole genome screens,
with increasing power suggest as indicated above that many of the prior loci
are either false positives, have such small effects that they were not detected
in the genome screens, or are related to only specific populations, as for
instance is suggested for the SUMO4 gene for only Asian patients 46. Table 7.2 summarizes “significant” regions
for the whole Wellcome Trust case control study using the combined “control”
reference population of 7,670 controls compared to 2,000 patients with type 1
diabetes (The locus for IFIH1 did not reach “significance” in this Wellcome
whole genome analysis with the SNPs analyzed, but is included in Table 7.2
related to a follow-up study29).
Table 7.2. Post-whole genome screens (2007) -Susceptibility
Loci for Type 1 Diabetes; Bolded loci <10(-7);
underline <10(-5) WTCCC analysis
|
Locus |
Chromosome |
Candidate
Genes |
Markers |
P
(-10)
|
Hetero
OR |
Homo
OR |
|
IDDM1 |
6p21.3 |
HLA DR/DQ
|
rs9272346 |
134 |
5.49 |
18.52 |
|
IDDM2 |
11p15.5 |
INSULIN VNTR
|
rs689;rs3741208 |
|
|
|
|
PTPN22 |
1p13 |
PTPN22 (LYP)
|
rs6679677Rs2476601=R620W |
41 |
1.82 |
5.19 |
|
IDDM12 |
2q33 |
CTLA-4 |
rs3087243 (AT)n
3 |
6 |
|
|
|
|
2q24 |
IFIH1 |
Rs1990760 |
3 |
|
|
|
|
10p15 |
IL2RA(CD25) |
rs2104286;rs52580101;rs11594656;
rs706778; D10S1426, D10S565 |
8 |
1.30 |
1.57 |
|
|
12q13 12q14-q12 |
?ERBB3 |
rs11171739, rs2292239 D12S375 |
11 |
1.34 |
1.75 |
|
|
3p21 |
|
|
7 |
|
|
|
|
12q24 |
?C12orf30,SH2B3,TRAFD1,PTPN11 |
rs17696736, rs3184504 |
14 |
1.34 |
1.94 |
|
|
16p13 (16p12-q11.1) |
KIAA0350 |
rs12708716 D16S3131 |
10 |
1.19 |
1.55 |
|
|
17q21 17q25 |
|
|
6 |
|
|
|
|
18p11 |
PTPN2 |
rs2542151;rs1893217; rs478582 |
7 |
1.30 |
1.62 |
|
|
18q22 |
?CD226 |
rs763361 |
|
|
|
|
|
22q13 |
?IL2RB |
rs229541 |
6 |
|
|
|
|
12p13 |
?CD69, CLEC |
rs11052552 |
8 |
1.57 |
1.48 |
The Major
Histocompatibility Complex, IDDM1
The major locus for type 1 diabetes susceptibility is
located within the HLA (Human Leukocyte Antigen) region 47
on the short arm of chromosome 6 48 and is calculated to provide up to 40-50% of the
inheritable diabetes risk 49, though
this calculation is based upon certain assumptions, including negligible
recombination between susceptibility loci in the region. We believe this assumption may not be valid,
if loci, far telomeric or centromeric of DR and DQ class II genes, contribute
to genetic susceptibility. The HLA
complex was first linked to diabetes when associations with several HLA class I
antigens (HLA-B8, -B18, and -B15) were discovered by serological typing and
affected sib-pairs showed evidence of linkage 50-52. With the development of novel typing
reagents, HLA class II genes (DQ, DR, and DP in that order of risk) were shown
to be even more strongly associated with the disease 52-54. However, several loci within or near the HLA
complex appear to modulate diabetes risk, and add further complexity to the
analysis of IDDM1-encoded
susceptibility 30, 55. Alleles, modes of
inheritance and putative mechanisms of susceptibility encoded for at the IDDM1 locus are discussed below. A schematic representation of the HLA region
and its association with IDDM is shown in Figure 7.4. A recent manuscript
analyzes a large number of families of the Type 1 Diabetes Genetics consortium
and documents the influence of multiple class
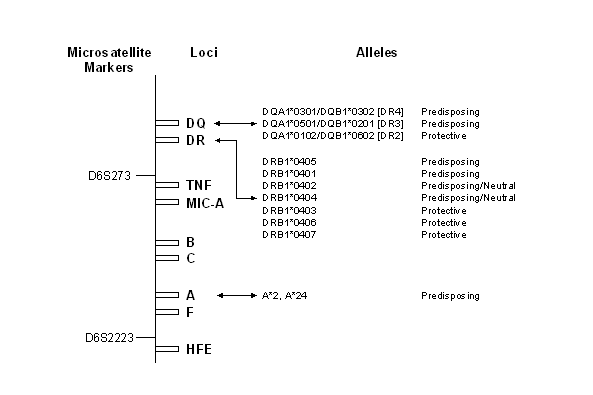
Figure 7.4 The HLA Region and
IDDM Susceptibility. Schematic
representation of the HLA region showing microsatellite markers, loci, and
alleles associated with IDDM susceptibility. Distances between loci are grossly
approximated.
The HLA Class II Region. The great majority of Caucasian patients have the HLA-DR3
or -DR4 class II alleles and approximately 30% to 50% of patients are DR3/DR4
heterozygotes 56.
The DR3/DR4 genotype confers the highest diabetes risk with a synergistic mode
of action, followed by DR4 and DR3 homozygosity, respectively 57 [See
attached teaching slides by J. Noble summary of HLA nomenclature]. Following
the development of DNA-based sequencing and typing technology, the HLA-DQ locus
was found to be the most strongly associated with diabetes susceptibility. This locus encodes for multiple variants of
the HLA-DQ molecule, a heterodimer consisting of two chains (a and ß)
involved in immune recognition and antigen presentation to CD4 T cells. In Caucasians, the HLA-DQ heterodimers (the
αchain genes are labeled DQA1 and the ß-chain genes DQB1) encoded by the
DQA1*0301, DQB1*0302 and DQA1*0501, DQB1*0201 alleles have the strongest
association with diabetes. These alleles are in linkage disequilibrium with the
HLA-DR4 and -DR3 alleles (Table 7.3), respectively 58. Linkage
disequilibrium often extends centromeric and telomeric of the class II region 59-61.
Table
|
HLA-DR |
DQA1 |
DQB1 |
DRB1 |
Susceptibility |
|
DR2 |
0102 |
0602 |
1501 |
Protective |
|
DR2 |
0102 |
0502 (AZH) |
1601 |
Predisposing |
|
DR2 |
0103 |
0601 |
1502 |
Neutral |
|
DR3 |
0501 |
0201 |
0301 |
High Risk |
|
DR4 |
0301 |
0302 |
0401 |
High Risk |
|
DR4 |
0301 |
0302 |
0402 |
Predisposing |
|
DR4 |
0301 |
0302 |
0403 |
Lower Risk |
|
DR4 |
0301 |
0302 |
0404 |
Predisposing |
|
DR4 |
0301 |
0302 |
0405 |
High Risk |
|
DR4 |
0301 |
0301 |
0401 |
Neutral |
|
DR4 |
0301 |
0303 |
0401 |
Neutral |
|
DR7 |
0201 |
0303 |
0701 |
Protective |
|
DR6 |
0101 |
0503 |
1401 |
Protective |
Allelic
variation at the DQB1 locus differentiates diabetes susceptibility among the
two most common HLA-DR4 haplotypes found in Caucasians based on the presence of
the DQB1*0302 or DQB1*0301 allele. Most
patients with DR4 carry the DQB1*0302 allele, while the DQB1*0301 and *0302
alleles are more evenly distributed in the general population. An independent effect has not been
demonstrated for DQB1*0201 because of the strong linkage disequilibrium between
DQB1*0201 and DRB1*0301 on Caucasian DR3 haplotypes. However, DQB1*0201 does not confer increased
susceptibility in association with DRB1*0701 on DR7 haplotypes. The different risk conferred by DQB1*0201
when on a chromosome with DR3 or DR7 may be explained by the different DQA1
alleles associated with DQB1*0201 on such haplotypes (DQA1*0501 on DR3,
DQA1*0201 on DR7) 62, 63. In addition, other susceptibility loci may be
in linkage disequilibrium with DQB1*0201 on DR3 haplotypes 49, although the class II region may be the
primary risk determinant on DR3 haplotypes 64. Trans-complementation of DQ α- and
ß-chains from opposite haplotypes has been demonstrated, and this significantly
increases the diversity of class II antigens participating in the immune
response and the potential for HLA-DQ contribution to IDDM susceptibility. A trans-complementing DQ molecule would be
unique to a heterozygous individual and usually it would not be expressed in
his parents. Thus, this phenomenon has
been proposed as an explanation for the increased diabetes risk observed in
DR4, DQA1*0301, DQB1*0302/DR3, DQA1*0501, DQB1*0201 heterozygotes 63.
(Erlich)
DQB1*0302 differs from DQB1*0301 at position 57, where it
lacks an aspartic acid residue, similar to the I-A molecule of the NOD mouse
(reviewed in ref. 65). The DQB1*0201 allele also lacks aspartic acid at position
57, and it has been proposed that this residue may be involved in the molecular
mechanism underlying IDDM1-encoded
susceptibility 58, 61, 61. In fact, the amino acid residue at position 57 of the DQ-ß
chain appears to be critical for peptide binding and recognition 66. Other residues of
the DQ-ß chain may influence peptide binding and diabetes susceptibility, and
in particular the combined variation of residues at positions 57 and 70 seem to
more strongly correlate with diabetes risk 67, 68, 68. An arginine residue
at position 52 of the DQ-α chain also correlates with diabetes
susceptibility 62. The importance of
the residue at position 57 has been disputed by trans-racial studies showing
that DQB1 alleles found with increased frequency in Japanese patients carry
instead of lack an aspartic acid residue at this position 69. Patients carrying similar Asp57 high risk alleles are also
found among Caucasians 70-79 (Figure 7.5). Moreover, certain low risk DQB1
genotypes also lack aspartic acid at position 57, including DQB1*0302/DQB1*0201 (DR7), and DQB1*0201
(DR3)/DQB1*0201 (DR7).
It is important to recognize that even the class II MHC genes with the greatest impact on diabetes susceptibility have a complex inheritance and their effect on risk cannot be explained by relatively simple rules (for instance, based on the presence of certain amino acid residues in the DQ genes). As illustrated below (Figure 7.5), the rule that lack of aspartic acid at position 57 of the DQB1 gene is strongly associated with risk is not consistent with relatively potent diabetogenic DQ alleles such as DQB1*0303 and DQB1*04 (usually 0401 in Caucasians and 0402 in Korean and Japanese patients) 80.
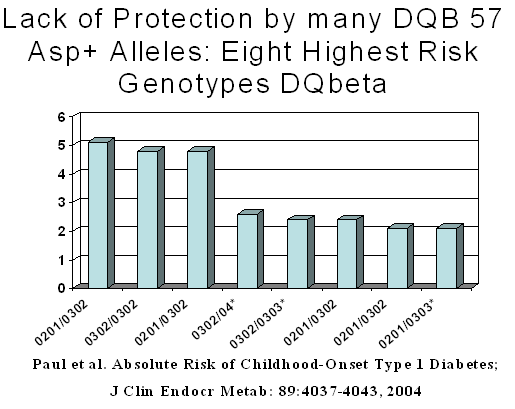
Figure 7.5
High-risk genotypes in population based
However, there is clear evidence that certain residues have
a functional role in determining binding and presentation of certain peptides 81. By using X-ray
crystallography, investigators have determined the three-dimensional structure
of the HLA-DQ8 molecule (encoded by DQA1*0301/DQB1*0302) complexed with an
immunodominant peptide of the insulin molecule (insulin B:9-23) 82. The DQ8 structure
suggests that the residue at position 57 contributes to the shaping of the P9
pocket, which together with the P1 and P4 pockets appear relevant to diabetes
susceptibility. The P4
pocket is deeper in DQ8 compared to DR1, DR2, DR3, DR4 but predictably similar
in HLA-DQ2 (DQA1*0501, DQB1*0201) and the diabetes protective HLA-DQ6
(DQA1*0102, DQB1*0602), thus not directly correlating with susceptibility. Moreover, the binding pockets of HLA-DQ8 were similar to
those of HLA-DQ2 and to those of the I-Ag7 molecule (corresponding to human
DQ), the main genetic susceptibility locus in NOD mice. This finding suggests that diabetes may
depend on antigen-presentation event(s) that may be similar in humans and NOD
mice. In further support of this hypothesis, it has been shown that HLA-DQ8
and I-Ag7 select common peptides, use the same binding register, which is not
promiscuous and is rather selective and dominated by the P9 pocket 83, 84. Though it
was generally assumed that autoantigenic peptides presented by high risk Asp 57
alleles (e.g. I-A87, DQ8, DQ2) would bind to the class II allele with a
negatively charged amino acid binding to pocket 9, our recent studies of major
insulin peptide B:9-23 and I-A27 of NOD mice indicates just the opposite. Namely the peptide, though it can bind in multiple
registers, is only recognized by diabetogenic T cell receptors when it binds
with B22 org amino acid projecting into pocket 9. This is a low affinity unfavorable binding
register and we hypothesize that T cells reacting with the B:9-23 peptide escape
thymic deletion because so little of the peptide binds in an appropriate
register to delete the relevant anti B:9-23 T cells. In contrast to the thymus, islets produce a
huge amount of insulin and the B:9-23 peptide, presumably allowing targeting of
islets
IDDM1-encoded susceptibility is mostly conferred by alleles of
the HLA-DQ locus in the class II region. The above conclusion is also supported
by the fact that the DQA1*0102, DQB1*0602 alleles, encoding for the HLA-DQ6
heterodimer found on HLA-DR2 haplotypes, confer dominant protection from the
development of type 1 diabetes (reviewed in ref. 85). Among four common
DR2 haplotypes observed in Caucasians, the DQA1*0102, DQB1*0602, DRB1*1501
haplotype is negatively associated with type 1 diabetes and is reported in less
than 1% of patients in most populations studied, including those of Caucasian
(both European and North-American) 75, 76, 82,
86-91, Asian 69, 92, 93, African-American 74, 75, and Mexican-American origin 94.
The DQB1*0602 allele in particular is the only class II
allele exclusively found on protective DR2 haplotypes while all the other
alleles (DQA1*0102, DQA1*0103, DQB1*0601, DQB1*0502, DRB1*1501, DRB1*1502,
DRB1*1601) can be found on neutral or moderately predisposing DR2
haplotypes. Moreover, a few rare
patients with type 1 diabetes have been described carrying mutated DQB1*0602
alleles or unusual DQA1/DQB1 alleles in cis with the usual DRB1*1501
allele. Thus, the available evidence
suggest that the diabetes-protective effect associated with DR2 haplotypes may
be mostly mapped within the DQ locus and in particular to the DQB1*0602
allele. Although a number of patients
with DQB1*0602 have been identified 91, the overall number is small (approximately 1% of children
developing diabetes and perhaps 5% of adults from the Swedish population) 95. Protection appears
to be dominant since DQB1*0602 protects from diabetes even in the presence of
high-risk HLA alleles 73, 86.
However, a subset of HLA-DR2, DQB1*0602 haplotypes marked by
alleles at the D6S265 locus has been identified as less protective (but still
markedly protective) in a Swedish cohort. HLA-DR2 (DRB1*15), DQB1*0602
haplotypes carrying D6S265*15 have a ten-fold higher odds ratio (OR) than those
carrying other alleles and thus confer reduced protection (OR with D65265*15
0.186 (.074 to .472) versus .017 (.005 to .062)). Marker
D6S265 maps 100 kb telomeric of the HLA-A
locus, which has been previously associated with diabetes
susceptibility. Associations between D6S265 and other autoimmune diseases have
been reported, including an association with multiple sclerosis and D6S265
specifically on HLA-DRB1*15, DQB1*0602 haplotypes 96. Thus, genetic variation at D6S265 can influence or
is linked to a locus that can influence susceptibility to or protection from
the autoimmunity conferred by HLA-DRB1*15,
DQB1*0602 haplotypes. Known genes that may be marked by D6S265 include HLA-A,
HLA-B, MICA, TNF and BAT1. Polymorphisms at these loci may have important
effects on the function of cytotoxic T cells and cytokine secretion. Moreover,
possible effects on transcriptional regulation may perhaps influence the
expression of the HLA-DQ molecule encoded by DQA1*0102, DQB1*0602. Further
characterization of this region will be needed to identify the loci that
contribute to the genetic protection from type 1 diabetes conferred by DRB1*15,
DQB1*0602.
Studies in transgenic mice have provided direct evidence
that the DQ locus, and the DQB1*0302 allele in particular, can engender an
immune response leading to the development of diabetes 74. However, the mechanism by which the HLA-DQ locus
influences diabetes susceptibility is the subject of intense speculation. Since
HLA-DQ molecules are known to play a role in antigen presentation, allelic
variation at this locus may affect the binding and functional properties of DQ
heterodimers and in turn the presentation of islet cell antigen-derived
peptides to immunocompetent cells.
Protective HLA molecules may have higher affinity for one or several
peptides than predisposing molecules. Therefore, it is suggested that
predisposing HLA molecules may be ineffective at binding and presenting
peptides derived from islet cell antigens. Indeed, the HLA-DQ molecules encoded
by the protective DQA1*0102, DQB1*0602 and predisposing DQA1*0301, DQB1*0302
alleles appear to differ in their affinity and specificity for peptides derived
from the insulin, glutamic acid decarboxylase (GAD), and tyrosine phosphatase
IA-2 autoantigens 76, 77, 77. Similar findings
were reported for DR molecules, with protective HLA-DR2 (DRB1*1501) molecules
displaying stronger affinity for (pro) insulin peptides than susceptible
HLA-DR3 (DRB1*0301) molecules 78. Our recent studies
of I-A87 suggest that binding in a low affinity register may be key to
autoreactivity to NOD target peptide B:9-23.
It is unclear whether genetically determined differences in
peptide binding and presentation affects the shaping of the T-cell repertoire
in the thymus or modulates immune responses in the extra-thymic periphery. A
poor presentation in the thymus could impair mechanisms of negative selection
allowing autoreactive T cells to escape deletion. In contrast, a protective
HLA-DQ molecule could promote tolerance to ß-cell molecules by eliciting more
efficient antigen presentation and negative selection in the thymus. Although
several studies involving the transgenic expression of MHC molecules in mice
did not support this hypothesis 79, 97, a study provided novel evidence for thymic deletion as a
mechanism of protection associated with MHC genes in transgenic mice 98. Moreover, the
demonstration that insulin and other islet cell antigens are ectopically
expressed in human thymus 88, 89 indirectly supports the hypothesis that thymic self-antigen
presentation and deletional mechanisms may be affected by the affinity and
binding properties of HLA-DQ and HLA-DR molecules.
An alternative hypothesis is that DQB1*0602-associated
protection may be mediated outside the thymus through the stimulation of
regulatory immune responses associated with peripheral tolerance. The predominance of Th2 responses is usually
associated with lack of progression to overt diabetes (reviewed in ref. 90) and regulatory T-cells are essential for the prevention of
autoimmunity. There is indeed evidence that a non-diabetogenic immune response,
mostly limited to the production of autoantibodies against the GAD autoantigen,
may occur in first degree relatives with DQB1*0602 73, and in whom the presence of DQB1*0602 and DQA1*0102 has
been confirmed by direct sequencing of the second exon 91. A similar response has been reported in patients with type
1 autoimmune polyendocrine syndrome who do not invariably progress to overt
diabetes 99, 100. Moreover, a similar protective effect has also been
reported in first degree relatives participating in the ongoing Diabetes
Prevention Trial (DPT-1), although different degrees of protection may occur in
different ethnicities 101. The presence of GAD
autoantibodies, often at high titers, may reflect the predominance of Th2
responses in relatives with DQB1*0602.
Finally, the two hypotheses are not mutually exclusive and
DQB1*0602-associated protection could be mediated both in the thymus and the
periphery.
Two additional strongly protective haplotypes are DRB1*1401,
DQA1*0101, DQB1*0503 and DRB1*0701, DQA1*0201, DQB1*0303. The DRB1*1401
haplotype is particularly interesting in that it is a HLA-DR allele with an
apparent lack of transmission to affected children as dramatic as for DQB1*0602
(Both DRB1*1401 and DQA1*0201/DQB1*0303 are relatively infrequent but strongly
protective) 102. (Fig. 7.6).
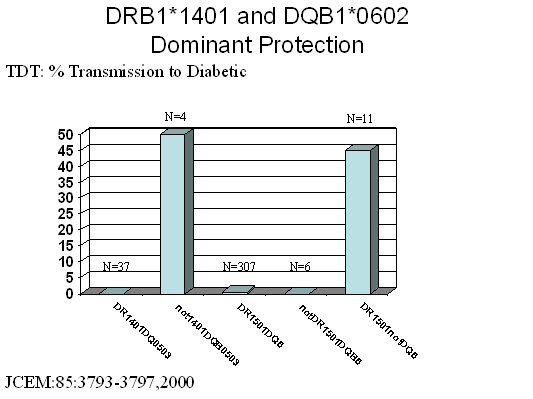
Figure 7.6: Transmission of DR/DQ haplotypes to patients
with type 1 diabetes. Note that DQ6
(DQB1*0602) containing haplotypes and DRB1*1401 containing haplotypes are
not/rarely transmitted to diabetics, while the usual DR or DQ alleles
associated are transmitted when DQB1*0602 (e.g. DRB1*1501) or DRB*1401
(DQB1*0503) are not present in the haplotype.
Other
loci in the class II region have been associated with diabetes susceptibility
besides HLA-DQ. Several studies indicate that DRB1 alleles (Figure 7.7)
significantly contribute and modulate diabetes susceptibility 60, 103-110. The DRB1*0405 and *0401 alleles have been reported as
predisposing, *0402 and *0404 as mostly neutral, while *0403, *0406, and *0407
appear to be protective.

Figure 7.7 Modified risk of diabetes relative
DRB1*04 alleles, with DRB1*0403/0403 even when combined with high risk
DQB1*0302, decreasing risk to background population (approximately 1/300).
There is evidence for contribution to risk from the DPB1
locus, confirmed in an extensive analysis by Valdes et al. 111-113. An independent
association has been observed in Mexican American 114 and Caucasians with DPB1*0301 115. The frequency of
DPB1*0101 is increased in patients (almost exclusively found on DR3
haplotypes). The maternal transmissions
of DRB1*0301-DPB1*0101 haplotypes to affected children occurred twice as
frequently as do paternal transmissions 49. Transmissions of
DR3 haplotypes carrying other DPB1 alleles occurred at approximately equal
maternal and paternal frequencies. A recent analysis indicates that the
DPB1*0402 allele, previously associated with decreased diabetes risk, is
associated with dominant protection from development of anti-islet autoantibodies
and diabetes in young children 38
amongst children having the highest risk DR3/4-DQ2/DQ8 genotype.
It is controversial whether loci (TAP1, TAP2) encoding for
peptide transporter genes associated with antigen processing and localized
centromeric to the DQ loci may also affect IDDM susceptibility 42, 116. Homozygosity for
the TAP2*0101 allele was associated with increased IDDM risk independent of
HLA-DQ susceptibility in a French study 117, but other studies have failed to show such independent
effect and suggested linkage disequilibrium between the HLA-DQ and TAP2 loci 118, 119. Of note, a mutation
at the same locus has been implicated as the cause of the class I deficiency
associated with IDDM in studies in humans and NOD mice 120-124.
The HLA Class
I Region. A number of
observations indicate that class II genes cannot explain all of the HLA
association with IDDM. A role for HLA
complex genes other than the DR-DQ or other class II genes was first
demonstrated by Robinson et al. 125. They examined affected sib pairs with parents homozygous
for the DR3 haplotype and used the HLA class I B locus to distinguish between
the two DR3 haplotypes of the homozygous parent. Under the null hypothesis that no HLA region
variation additional to that defined by the DR3 haplotype is involved in IDDM,
the affected sib pairs should share the two parental DR3 haplotypes at the same
frequency. Significant deviation from
50% sharing was observed. Since the DR3
haplotypes examined in this study could be assumed to be homogeneous for their
DR-DQ alleles at the molecular level (DRB1*0301 DQA1*0501 DQB1*0201), this test
implicated other HLA loci in IDDM susceptibility. Several other reports suggest that HLA class
I genes, and in particular the HLA-A24 allele, may also influence
susceptibility and particular clinical aspects of the disease such as age of
onset 126, 127 and the rate of
ß-cell destruction 98, 128-132.
Besides HLA-A24 (*2401), other class I alleles are independently associated
with susceptibility (HLA-A*0101 and *3002) and though uncommon B3906 is
strangely associated with diabetes risk. 133. There is also evidence that several alleles at the
class I HLA-B and C loci modulate susceptibility and influence age of onset 134. A risk-modifying locus may lie between HLA-B and
marker D6S2702, which is located 970 kb telomeric of HLA-B 135.
Another diabetes-associated locus has been found in the
class I region, telomeric to HLA-F 42. By considering the transmission ratios of microsatellite
variation from parents homozygous for the HLA class II DR-DQ genes (using the
Homozygous Parent Transmission Disequilibrium Test), the possible confounding
effect of linkage disequilibrium was removed.
Evidence for a second IDDM locus in this region was demonstrated, near
the HFE (hemochromatosis) gene and 8.5 Mb distal to the HLA class II loci.
Analyses from three independent family data sets from
The HLA Class
III Region. Moghaddam et al. 118 analyzed 11 markers in the HLA region in IDDM patients and
controls fully matched for the highest risk DQA1*0501, DQB1*0201/HLA-DQA1*0301,
DQB1*0302 (DR3/DR4) genotype. Their
study provided strong evidence that another critical region for IDDM
susceptibility, approximately 200 kb in size, lies around the microsatellite
locus D6S273 which is located between the TNF and HSP70 genes. Another study has independently confirmed
linkage with marker D6S273 showing evidence for non-random transmission from
DRB1*03-DQA1*0501-DQB1*0201 homozygous parents 42. Further studies in multiplex families from the
In addition, the class I chain-related MIC-A and MIC-B
genes, located between the HLA-B and the TNFa genes, may also affect IDDM
susceptibility. MIC-A
polymorphisms are associated with disease susceptibility in several populations
138-145. In a case-control study of Italian patients, the frequency
of the MIC-A5 allele was increased in patients while none of the TNFa alleles
were statistically significantly associated with the disease. In this study, the MIC-A5 allele was
associated with IDDM independently of class II alleles, suggesting an
independent contribution of this locus to diabetes risk 138. MIC-A alleles have a strong effect on development of
Addison’s disease and a weaker apparent influence on the development of type 1
diabetes. However, homozygosity
for the MIC-A 5.1 allele (with a premature stop codon) was associated with
increased diabetes risk and faster progression to diabetes in young children
followed from infancy in the DAISY study, especially in those with the
HLA-DR3-DQ2/DR4-DQ8 genotype 146. Finally,
HSP70-2 and HSP70-Hom genes are also located in the class III region although
there is no evidence for an independent association with IDDM from studies that
could not circumvent linkage disequilibrium 126, 147-151.
Clinical Heterogeneity of Type 1
diabetes in Relation to the IDDM1 Locus.
Age dependent HLA heterogeneity has been observed in
Caucasian IDDM patients, indicating that high risk HLA genotypes occur at a
higher frequency among the younger age onset groups 75, 152-154, whereas older age at diagnosis is associated with an
increased heterogeneity of DRB1 and a decreased heterogeneity of DPB1 106. Caillat-Zucman and
coworkers have found a decreased frequency of DR3 and DR4 haplotypes and of
DR3/DR4 heterozygosity amongst patients who had developed diabetes after age 15
128. Similar findings were reported by Tait et al. 112. Earliest development of diabetes is strongly associated
with the DR3/DR4-DQ8 genotype and such a genotype is preferentially followed in
the population based DAISY (Diabetes Study of the Young) study 155.
The immunogenetic analysis of islet cell antibody (
Extended (Ancestral) MHC
Haplotypes
Early
studies by Alper and co-workers and Dawkins and co-workers documented the
existence of haplotypes in the MHC region that were very large and relatively
common where all polymorphic markers were essentially all conserved 162, 163. With high throughput SNP analysis or
extensive sequencing the remarkable size and conservation of these haplotypes
has been confirmed 164, 165. In
particular, the HLA-A1, B8, DR3 haplotype is often conserved for more than 2.7
megabases, within which greater than 99.9% of SNPs or sequences are
identical. Another remarkable haplotype
is the A30, B18, DR3 “Basque” haplotype 166. Overall the 8.1 haplotype does not show
enhanced transmission to diabetics compared to non 8.1 DR3 haplotypes 64 while the
HLA-A30, B18, DR3 haplotype does show enhanced transmissions. It is likely that careful analysis of these
haplotypes will aid in localization of additional genes contributing to type 1
diabetes susceptibility.
We have
evidence from analysis in the DAISY study that there is a major gene linked to
DR-DQ such that for siblings with DR3-DQ2, DR4-DQ8 sharing both MHC haplotypes
with their proband the risk of islet autoimmunity exceeds 60% 30 with type
1 diabetes following the appearance of islet autoantibodies by several years in
this high risk young population. In
contrast siblings with the same HLA DR and DQ alleles, but sharing only one or
no HLA haplotypes (despite being DR3/4-DQ2/DQ8) have a risk of activating
anti-islet autoimmunity of “only” 20% (figure 7.8). This strongly implicates non-DR/DQ loci, linked
to or within the major histocompatibility complex contributing to diabetes
risk. It also suggests that for this
DR-DQ genotype, environmental factors essential to activate anti-islet
autoimmunity are unlikely to be rare, given the extremely high penetrance of
disease.
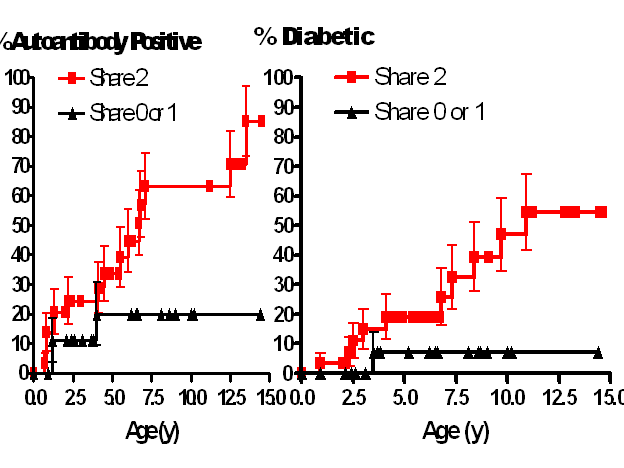
Figure
7.8 Extreme risk of type 1A diabetes for
siblings of patients with type 1 diabetes who share both HLA haplotype
identical by descent with their sibling proband 30.
The Insulin Gene Locus, IDDM2
Insulin was the only autoantigen in humans for which expression within
the pancreatic islet is specifically restricted to ß-cells. Recently
The insulin gene (INS) is therefore an obvious
candidate susceptibility locus. Its role in disease susceptibility was easily
demonstrated by association studies and was replicated by linkage analysis 45, 189. Indeed, the
4.1 Kb region containing INS
and its flanking regions contain several polymorphisms in linkage
disequilibrium that have been associated with diabetes risk 190.
Extensive studies
involving polymorphisms in the neighboring HUMTHO1
(tyrosine hydroxylase) and IGF2 genes
provided strong evidence that INS is the main susceptibility determinant
in this region 167, 168,
191-194. All of the
polymorphisms lie outside coding sequences, confirming that diabetes
susceptibility must derive from modulation of INS transcription.
Susceptibility in the INS region, or the IDDM2 locus, has been primarily mapped to a variable number of
tandem repeats (VNTR) located ~0.5 kb upstream of INS 195-197 (Figure
7.8). The VNTR may not explain all of the susceptibility in this region 182, 198 and
at least two other polymorphisms (-23HphI and +1140A/C) may
contribute to the etiological effect 199 (Figure 7.8).
The VNTR.
This polymorphic repeat, also known as the insulin gene minisatellite or ILPR
(insulin-linked polymorphic region), consists of a 14-15 bp unit consensus
sequence (ACAGGGGTCTGGGG) with slight variations of the repeat sequence. Any number from 30 to several hundred repeats
has been observed, but allele frequencies tend to cluster in the 30-60 repeats
range (class I alleles) or at 120-170 repeats (class III alleles). The
intermediate class II alleles are rare in Caucasians, and less rare in
individuals of African descent 200, 201. The
sequence of the VNTR is particularly G-rich, and it tends to form unusual DNA
structures in vitro and in vivo, presumably through the
formation of G-quartets 135, 202. Shortly
after its discovery 195, the insulin VNTR was found to be associated with type 1
diabetes 181. Homozygosity for
the short class VNTR I alleles is found in ~75-85% of the patients compared to
a frequency of 50-60% in the general population, suggesting that it predisposes
to type 1 diabetes. In contrast, homozygosity for the longer class III VNTR
alleles is rarely seen in patients and the class III VNTR is believed to confer
a dominant protective effect 181, 203, 204. The
relative risk ratio of the I/I genotype vs. I/III or III/III has been reported
to be moderate (in the 3-5 range) and it accounts for about 10% of the familial
clustering of type 1 diabetes 205. Moreover,
by measuring the HphI polymorphism
(in tight linkage disequilibrium with the VNTR) 190, Metcalfe
et al. 206 showed
that homozygosity for the predisposing INS
genotype increases the likelihood that identical twins will be concordant for
the development of autoimmunity and diabetes in the BabyDiab study, in which
offspring of affected parents are followed prospectively from birth (Fig. 7.9) 207. Halminen et al. 208 reported
that IDDM2-encoded susceptibility is
associated with reduced insulin secretory capacity found in
autoantibody-positive first-degree relatives (siblings) from the Childhood
Diabetes Study in
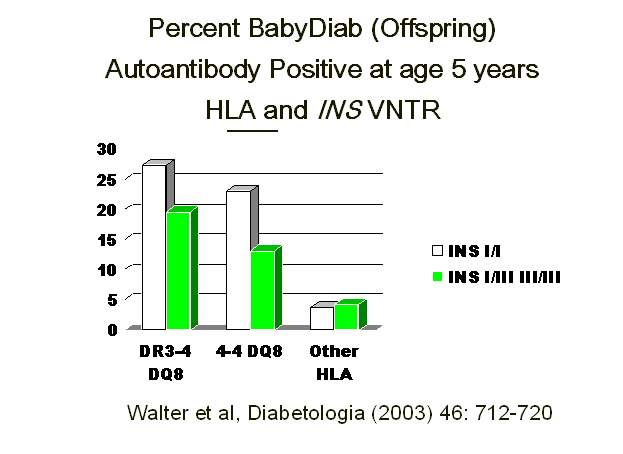
Figure 7.9
Insulin gene VNTR (Variable Nucleotide Tandem Repeats) polymorphisms increase
risk of developing anti-islet autoimmunity in the BabyDiab study.
VNTR heterogeneity. Although
VNTR alleles cluster in two main classes with divergent associations with type
1 diabetes, there is evidence that VNTR alleles are quite heterogeneous and may
differ in their ability to modulate disease susceptibility. Further classification of VNTR alleles is
indeed possible according to size differences, and at least 21 class I and 15
class III VNTR alleles were described by fluorescence-based DNA fragment sizing
technology 209 (Fig.
7.7). Bennett et al. grouped the 15
class III VNTR alleles identified according to two main modes of transmission
based on the linkage disequilibrium pattern with alleles at the HUMTHO1 locus on chromosome 11p15. Thus, by taking both size and flanking
haplotypes into account class III VNTR alleles linked to the HUMTH01 Z-8 allele were found more
protective (very protective haplotype or VPH) than those linked to the HUMTH01 Z allele (protective haplotype
or PH) (Fig. 7.7) 209, 210. However, certain VNTR alleles can be found in linkage
disequilibrium with either the Z or Z-8 alleles. The variable degree of protection observed
for these alleles may also be influenced by sequence heterogeneity and its
effects on the VNTR physical state and transcriptional activity 135, 202,
205, 209, 211. Sequencing studies have indeed identified several variants
of the commonest VNTR repeat sequence that characterize yet another level of
heterogeneity 195, 200, 201,
205, 212.
Studies have also analyzed the variant repeat distribution
within the VNTR using minisatellite variant repeat mapping by PCR (MVR-PCR) 213. Some
of the variation within the repetitive sequence most probably arises from
mitotic replication slippage at an estimated frequency of 10-3 per
gamete. However, sperm DNA analysis
revealed a second class of mutation occurring at a frequency of approximately 2
x 10-5 that involved highly complex intra- and inter-allelic
rearrangements which are probably meiotic in origin 214. These
events may help explain the heterogeneity of the VNTR locus. The combined
analysis of the variant repeat distribution and of the haplotypes flanking the
VNTR has allowed defining five new ancestral allele lineages 193. By
this approach, class III VNTR alleles can be divided into two diverging
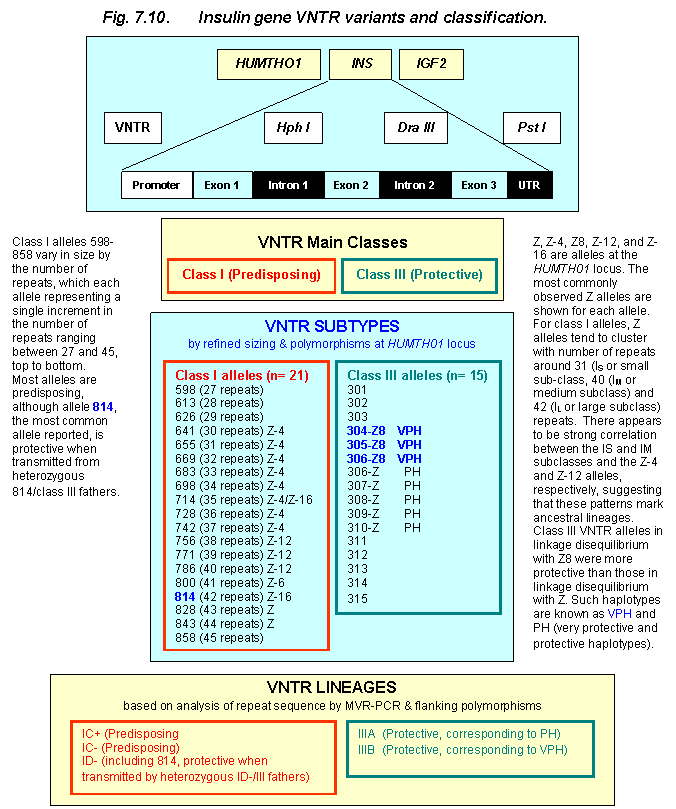
lineages, IIIA and IIIB (Fig. 7.10).
These two lineages correspond to the PH and VPH haplotypes previously
defined by Bennett et al. 209. Class
I alleles can also be divided into three newly defined lineages, IC+,
In contrast,
Figure 7.10 The IDDM2 Susceptibility Locus. Top to
bottom, the figure shows the HUMTH01, INS and IGF2 loci,
as well as a schematic structure of the insulin gene with the approximate
location of some of the most characterized polymorphic loci (VNTR, HphI, DraIII, PstI). Also shown are a schematic representation of
the two main VNTR classes, their association with diabetes, as well as the VNTR
alleles and allele lineages that have been defined with the variety of
approaches described in the figure and in the main text.
VNTR effects on transcription. Several studies have investigated the effects of the VNTR on
INS transcription. Transfection of rodent ß-cell lines with
reporter constructs representing the INS
promoter flanked by class I or class III alleles resulted in three-fold
differences going in opposite directions in reports from different laboratories
196, 197. These
discrepant results may be due to species-specificity, differences among
specific alleles within each class, or the absence of the genomic context
necessary for the VNTR to have its physiologic effects. Studies on the transcriptional effects of the
VNTR in vivo produced more meaningful
results. In fetal pancreas RNA, the INS transcript in cis with the class III VNTR was expressed at lower levels (15-20%)
than the class I transcript, a small but statistically significant difference 192. Bennett et al. 209 found
a somewhat larger difference in adult pancreas.
Moreover, single nucleotide differences in the VNTR sequence can affect INS transcription and correlate with the
ability to form unusual DNA structures, both at the inter- and intra-molecular
levels 211. These
findings led to the hypothesis that VNTR variants may differ in their ability
to stimulate transcription as a function of the binding of inter- and
intra-molecular quartets with the transcription factor Pur1. However, the transcriptional activity of
these variants observed in vitro may
not always correspond to that in vivo,
where overall transcription may depend on the interaction with other proteins
involved in the transcriptional machinery and on differences among the various
cell types that actively transcribe the insulin gene. The studies described
above report only marginal differences in pancreatic INS transcription, and the lower transcription associated with
diabetes-protective class III VNTR alleles does not fit well with their
dominant protective effect. It seems
unlikely that such minor differences in pancreatic INS transcription may
influence susceptibility to a form of diabetes resulting from the autoimmune
destruction of pancreatic ß-cells.
It was later discovered that INS is actively transcribed in the thymus in mouse 198, rat 215, and
humans 88, 89. The
human thymus was found to express low levels of INS message throughout fetal development and childhood but also
during adulthood 203. Overall,
genes encoding for several self-molecules have been found to be expressed in
the thymus, including pancreatic and thyroid hormones, neuroendocrine molecules
and other peripheral proteins 216. Functional studies in transgenic
mice and fetal organ thymic cultures have provided both in vivo and in vitro data
showing that thymic expression of self-antigens and their levels of expression
can dramatically affect the development of self-tolerance (reviewed in 217). The fact that negative selection of autoreactive thymocytes
is dose-dependent suggested the hypothesis that different VNTR alleles may
modulate tolerance to insulin by affecting insulin expression levels in the
thymus. Consistent with this hypothesis,
INS mRNA levels in the thymus were
found to correlate with VNTR alleles in opposite fashion to that observed in
the pancreas 209. INS transcripts
in cis with class III VNTR alleles
are transcribed at much higher levels (on average 2-3 fold) than those in cis with class I VNTR alleles 88. The
increased transcription levels detected in thymus fit well with the dominant
protective effect associated with class III VNTR alleles, as higher insulin
levels in the thymus may more efficiently induce negative selection of
insulin-specific T-lymphocytes (or improved selection of regulatory T
cells). In contrast, homozygosity for
diabetes-associated class I VNTR alleles determines lower insulin levels that
may be associated with a less efficient deletion of insulin-specific
autoreactive T-cells (or impaired selection of regulatory T cells). Proinsulin appears to be the main product of
the insulin gene in the thymus 89, 203. This
is not surprising since thymus cells expressing proinsulin are not likely to
possess the refined machinery necessary to process proinsulin to mature
insulin. Proinsulin expression may be
sufficient to obtain tolerance to insulin since most of the known immunodominant epitopes identified as targets of the
insulin autoimmune responses in type 1 diabetes are shared by both insulin and
proinsulin. Both thymic epithelial cells and
bone marrow derived dendritic cells have been shown to transcribe INS
and other genes coding for self-molecules 203, 217-221. Similar
cells and INS transcription have also
been demonstrated in peripheral lymphoid organs,
suggesting that insulin expression in lymphoid organs may also play a role in
maintaining peripheral self-tolerance throughout life 203, 222. Direct
support for the hypothesis that levels of INS
expression in thymus and lymphoid organs can influence type 1 diabetes
susceptibility is provided by studies in insulin gene knockout mice and
transgenic mice 183, 185,
223-225, which were recently reviewed 217.
Other effects at the IDDM2 locus. It is also possible that other loci in the
11p15 region may also contribute to IDDM2-encoded
susceptibility, as suggested for certain class I VNTR allele lineages (
Overall, the studies reviewed here suggest that
the IDDM2 is a quantitative trait
resulting from allelic variation and, as discussed in a later paragraph, from
complex parental and epigenetic effects at the VNTR locus. IDDM2-associated susceptibility and resistance may derive from
quantitative differences in INS
transcription in the specialized antigen presenting cells found in thymus and
peripheral lymphoid tissues, where production of self-antigens such as
proinsulin may be crucial for the shaping and maintaining of a self-tolerant T
cell repertoire 203, 235, 236. Such mechanisms may influence the probability
of developing autoimmune responses to insulin, a key autoantigen in type 1
diabetes.
PTPN22 (Lyp)
This gene was also identified through the
candidate gene approach. Bottini and coworkers evaluated a functional
polymorphism in the lyp gene (no relation to the lymphopenia gene of the BB
rat) in two series of patients with type 1 diabetes, one from
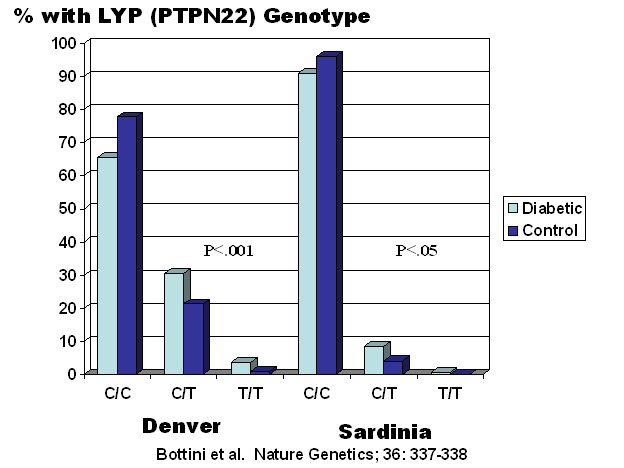
Figure 7.11 PTPN22 (Lyp)
genotypes. The minor T allele is associated with type 1 diabetes.
CTLA-4
(IDDM12)
Linkage with markers on chromosome 2q33 was initially
reported in a group of Italian families 238. This chromosomal
region contains the CTLA-4 (cytotoxic T lymphocyte associated-4) and CD28
genes, which encode for two molecules that are intimately involved in the
regulation of T-cell activation and proliferation. Differential regulation of these molecules
could easily affect T-cell function and hence the regulation of immune
responses. The CTLA-4 gene is a strong
candidate gene for autoimmune diseases since it encodes for a molecule that
functions as a key negative regulator of T-cell activation, and the linked
markers encompass a region containing an (AT)n microsatellite located in the 3
Further confirmation of association with the IDDM12-CTLA-4 locus came through linkage
disequilibrium (association) analysis using a multi-ethnic c
A multiethnic (U.S. Caucasian, Mexican-American, French,
Spanish, Korean, and Chinese) c
In a very large combined analysis of more than 3,600
families, Ueda and coworkers reported that a CTLA-4 polymorphism was
transmitted to 53.3% (versus the expected transmission of 50%) to affected
individuals, with a relative risk 1.14 (figure 7.10) 249. Susceptibility was mapped to a polymorphism
in the non-coding 6.1 kb 3’ end associated with lower messenger RNA levels of a
soluble form of CTLA-4, which results from alternative splicing. The 49 exon 1
G/G variant is associated with decreased expression of a soluble variant of
CTLA-4 that may have an influence on immune function, especially in light of
CTLA-4 polymorphism associated with diabetes of the NOD mouse 250, Graves’ disease 251, and Addison’s disease 252. Evidence for
linkage was also obtained in 2005 scan of the T1DGC, albeit the region is
likely to contain also IDDM7 45. Because the
effect of the reported CTLA-4 polymorphism in human diabetes is so small, lack
of confirmation in smaller studies or discordant results among studies is to be
expected 251. However, the biological contribution of this polymorphism
remains to be assessed by functional studies. Overall, CTLA-4 appears to be a
stronger determinant for Graves’ disease than for type 1 diabetes (figure
7.12). Concordant with its relatively
weak effect in the Wellcome Trust Case Control Consortium genome-wide association
study of 2,000 patients and 3,000 controls, no SNPs at 2q33 were significant at
either the 5X10(-7) cutoff27. Of note, CTLA4 G allele of rs3087243 was more
strongly associated with patients with type 1 diabetes plus thyroid peroxidase
autoantibodies (OR=1.49) compared to those without the thyroid autoantibodies
(OR 1.16)253.

Figure 7.12 Summary of CTLA-4
association with type 1 diabetes (man and mouse) and Grave’s disease.
IL2RA(CD25)
In the
Wellcome Trust Case Control Consortium Study SNP rs2104286 was analyzed with
rs706778 referenced as the prior reported SNP (HApMAp r2=.25) with
finding of an association trend value of 8.0X10(-6), and p value of
approximately 10(-8) with expanded control series. The odds ratio was 1.30 for heterozygotes and
1.57 for homozygotes 27. There
is evidence of two different SNPs associated with type 1A diabetes for the
IL2RA region (ss52580109 (P=7.8X10(-11)) and rs11597367(P=8.19X10(-7)). The odds ratio for the minor A allele of
rs11597367 was 0.78 (1.0/0.78=1.28). The
odds ratio for the ss52580109 minor A allele was 0.68 (1/0.68=1.47)254. The
associated SNPs are located in region of intron 1 of IL2RA and 5’ intergenic
sequence between IL2Ra and RBM17 (RNA binding motif protein 17). There is an interesting observation (Figure
7.13) that soluble IL2 receptor correlates with the genotype of the SNPs,
though with very extensive overlap, leading to the hypothesis that influence on
diabetes might relate to lower immune responsiveness contributing to type 1
diabetes 254. The
association of the two different SNPs appear to have different relations to
soluble IL2RA by genotype.
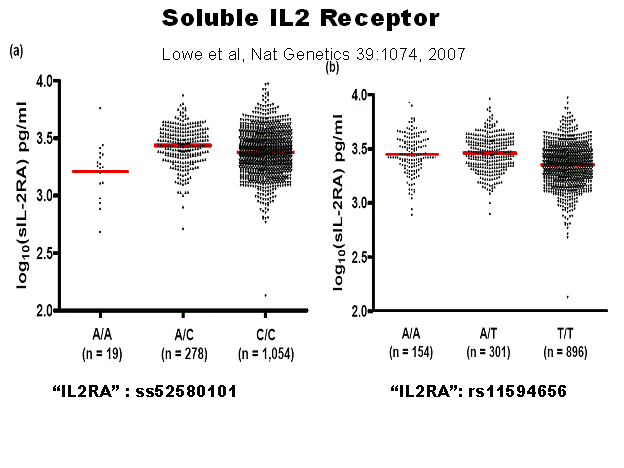
Figure 7.13
Soluble IL2 receptor levels (serum) relative to IL2RA SNP
genotypes. Lowe et al, Nature Genetics
39:1074, 2007.
IFIH1
The minor allele of rs1990760 of the Interferon Induced Helicase
region (IFIH1) was reported to be associated with type 1 diabetes with a risk
ratio of 0.86 in a large study of 4,253 cases, 5,842 controls, and with an
additional 2,134 parent-child trio analysis. The Wellcome Trust analysis
utilizing a different SNP (rs3788964) found a genotypic P-value of 7.6X10(-3)
and Trend p-value of 1.9X10(-3), suggesting a very modest association in this
study 255. The
gene is of particular interest in that it may relate the innate immune system
to the development of a disease presumably mediated by the adaptive immune
system, and animal models are available where activation of innate immunity,
and interferon alpha, is associated with induction of autoimmune diabetes 256, 257.
KIAA0350 (16p13)
The
association of KIAA0350 with type 1 diabetes was discovered with the Wellcome
Trust Case Control Consortium study with a Trend P Value of 9.2X10(-8) and a
heterozygous odds ratio of 1.19 and homozygous of 1.55 (SNP rs12708716) 27. The
association was confirmed with analysis of 4,000 patients and 5,000 controls
(10(-8)) and in a family analysis (trios, 10(-6)). There are only two genes in the region, the
lectin KIAA0350 and dexamethasone-induced transcript. KIAA0350 is a putative C-type lectin, and
also has an immunoreceptor tyrosine-based activation motif (ITAM) 29.
PTPN2 (18p11)
In the Wellcome Trust the region associated with PTPN2
(protein tyrosine phosphatase, non-receptor type 2) was associated with all of
the autoimmune disorders studied, namely Crohn’s disease, rheumatoid arthritis
and type 1 diabetes (for type 1 diabetes P=1.9X10(-6) with a heterozygote odds
ratio of 1.30 and homozygote odds ratio of 1.62 27.
Follow up study gave a P value of 3.36X10(-10) with an odds ratio of
1.29 27. The molecule
is a member of the same family as PTPN22, an allele of which is strongly
associated with type 1 diabetes (R620W)27, 34.
The Wellcome Trust Case Control
Consortium (WTCCC) primary genome-wide association (GWA) scan 27 on seven diseases, including the multifactorial autoimmune disease type
1 diabetes (T1D), has recently shown associations at P < 5 ![]() 10-7
between T1D and six chromosome regions: 12q24, 12q13, 16p13, 18p11, 12p13 and
4q27. Four of those regions have been replicated by another big study including
4,000 individuals with T1D, 5,000 controls and 2,997 family trios 29, 30: there was strong evidence for
disease association for chromosomes 12q24, 12q13, 16p13 and 18p11 in independent
cases and controls (P
10-7
between T1D and six chromosome regions: 12q24, 12q13, 16p13, 18p11, 12p13 and
4q27. Four of those regions have been replicated by another big study including
4,000 individuals with T1D, 5,000 controls and 2,997 family trios 29, 30: there was strong evidence for
disease association for chromosomes 12q24, 12q13, 16p13 and 18p11 in independent
cases and controls (P ![]() 1.82
1.82 ![]() 10-6),
in families (P = 5.23
10-6),
in families (P = 5.23 ![]() 10-3
to 1.07
10-3
to 1.07 ![]() 10-6)
and overall (P = 1.15
10-6)
and overall (P = 1.15 ![]() 10-14
to 1.52
10-14
to 1.52 ![]() 10-20).
10-20).
12q13
In the WTCCC,
SNP rs11171739 showed strong evidence of association with T1D with heterozygote
odds ratio (OR) of 1.34 and a homozygote OR of 1.75 and a genotypic P value of
9.71 x 10-11. This SNP rs11171739 is close to the ERRB3 gene (v-erb-b2
erythroblastic leukemia viral oncogene homolog 3) and another SNP rs2292239, in
the ERRB3 gene, has also shown association with T1D in the study by Todd et al 29, 30. The
ERRB3 gene encodes a member of the epidermal growth factor receptor (EGFR) family
of receptor tyrosine kinases. Amplification of this gene and/or overexpression
of its protein have been reported in numerous cancers, including prostate,
bladder 258 and
breast tumors 259. In
diabetic rats, expression of both ERRB2 and ERRB3 is enhanced during oral
oncogenesis, possibly resulting in promotion of cell proliferation and
inhibition of apoptosis 260.
The VDR gene is located on chromosome 12q12-q14. Four common
single nucleotide polymorphisms (SNPs) in the VDR gene have been
studied: FokI T>C (rs10735810), BsmI A>G (rs1544410), ApaI
G>T (rs7975232), and TaqI C>T (rs731236). FokI
polymorphism in exon 2 results in an alternative transcription initiation site,
leading to a protein variant with 3 additional amino acids 261. SNPs BsmI and ApaI
are located in intron 8, and TaqI is a silent SNP in exon 9.
Several studies reported association of type 1 diabetes with one of
these four SNPs. Pani et al. genotyped 152 Caucasian families for these
four polymorphisms and suggested an association with T1D susceptibility in
Germans 262. Guja et al studied 204 Romanian
diabetic families and found that VDR FoqI F allele seemed to be predisposing
while TaqI T allele seemed to be protective 263. However, these associations have
not been confirmed in more recent and bigger studies: one study in the Finish
population (1000 cases and 2000 controls) 264, another report by Todd et al. with
up to 3,763 T1D families from the UK, Finland, Norway, Romania and US and 3414
case-control subjects from the UK 265, and finally a recently conducted
meta-analysis also found no evidence of association 266.
Recently, several studies have reported associations of type
1 diabetes and other autoimmune diseases with polymorphisms in the CYP27B1 gene
on chromosome 12q13.1-q13.3, which encodes 1[alpha]-hydroxylase, the enzyme
that converts 25-hydroxyvitamin D (25OHD3) into 1,25-dihydroxyvitamin D (1,25diOHD3).
Lopez et al. 267 report
a significant association between allelic variation of the promoter (-1260) C/A
polymorphism and Addison
Lower serum concentrations of 1,25-dihydroxyvitamin D
(1,25diOHD3), the hormonally active form of vitamin D, and of its precursor
25-hydroxyvitamin D (25OHD3) have been reported at the diagnosis of type 1
diabetes compared with normal control subjects 269,
270. Epidemiological studies have
suggested that vitamin D supplementation in early childhood is associated with
a decreased risk of developing type 1 diabetes 271,
272. In the immune system, vitamin D
and its analogs have been shown to function by stimulating Th-2
T-helper cells to produce transforming growth factor-![]() 1
and IL-4 that might serve to suppress the TNF-
1
and IL-4 that might serve to suppress the TNF-![]() and interferon-
and interferon-![]() production by Th-1 cells 273. In the animal models, 1,25diOHD31
and its analogs have been effective in prevention diabetes in NOD mice 274,
275.
production by Th-1 cells 273. In the animal models, 1,25diOHD31
and its analogs have been effective in prevention diabetes in NOD mice 274,
275.
12q24
The SNP (from
the WTCCC study) rs17696736 in C12orf30 (Chr. 12 open reading frame 30) maps to
regions of extensive linkage disequilibrium covering more than ten genes.
Several of these represent functional candidates genes because of their
presumed roles in immune signaling, considered to be a major feature of
T1D-susceptibility. These include SH2B3/LNK (SH2B adaptor protein 3), TRAFD1
(TRAF-type zinc finger domain containing 1) and PTPN11 (protein tyrosine
phosphatase, non-receptor type 11). In the study by Todd et al. 29, 30,
rs3184504, a nsSNP in exon 3 of SH2B3 encoding a pleckstrin homology
domain (R262W), had the highest association (P = 1.73 ![]() 10-21;
OR = 1.33, 95% CI = 1.26–1.42). SH2B3 is an adaptor protein that regulates
growth factor and cytokine receptor-mediated pathways implicated in lymphoid,
myeloid and platelet homeostasis. It has been shown to negatively regulates
TNF-alpha expression in endothelial cells 276.
TRAFD1, also known as FLN29, is a novel interferon- and LPS-inducible gene that
acts as a negative regulator of toll-like receptor signaling 277. PTPN11 is probably the most attractive
candidate gene in the region given a major role in insulin and immune signaling
278. It is
also a member of the same family of regulatory phosphatases as PTPN22,
already established as an important susceptibility gene for T1D and other
autoimmune diseases.
10-21;
OR = 1.33, 95% CI = 1.26–1.42). SH2B3 is an adaptor protein that regulates
growth factor and cytokine receptor-mediated pathways implicated in lymphoid,
myeloid and platelet homeostasis. It has been shown to negatively regulates
TNF-alpha expression in endothelial cells 276.
TRAFD1, also known as FLN29, is a novel interferon- and LPS-inducible gene that
acts as a negative regulator of toll-like receptor signaling 277. PTPN11 is probably the most attractive
candidate gene in the region given a major role in insulin and immune signaling
278. It is
also a member of the same family of regulatory phosphatases as PTPN22,
already established as an important susceptibility gene for T1D and other
autoimmune diseases.
12p13
In the
multilocus analysis of the WTCCC, there was increased support for a region on
chromosome 12p13 containing several candidate genes, including CD69
(CD69 antigen (p60, early T-cell activation antigen)) and multiple CLEC
(C-type lectin domain family) genes. The SNP rs3764021 is located in the CLEC2D
(C-type lectin domain family, member D) gene, also known as LLT1 (lectin-like
transcript). The LLT1 receptor induces IFN-gamma production by human NK cells 279. CD69
is involved in lymphocyte proliferation and functions as a signal-transmitting
receptor in lymphocytes, NK cells and platelets. CD69 appears to be the
earliest inducible cell surface glycoprotein acquired during lymphoid
activation. Locus 12p13 has not been replicated so far.
22q13
SNP rs229541
close to the IL2RB gene shows evidence of T1D association in the WTCCC study (P
= 2.18 ![]() 10-6), but has not been replicated so far. The
IL-2 receptor, which is involved in T cell-mediated immune responses, is a
trimeric molecule of alpha (IL2RA), beta (IL2RB, also known as CD122) and gamma
(IL2RG) chains. IL2RB is an interesting candidate as IL2Ra on chr. 10p15 is
already a known susceptibility gene for T1D. IL-2 plays a major role in the
proliferation of cell populations during an immune reaction 280.
10-6), but has not been replicated so far. The
IL-2 receptor, which is involved in T cell-mediated immune responses, is a
trimeric molecule of alpha (IL2RA), beta (IL2RB, also known as CD122) and gamma
(IL2RG) chains. IL2RB is an interesting candidate as IL2Ra on chr. 10p15 is
already a known susceptibility gene for T1D. IL-2 plays a major role in the
proliferation of cell populations during an immune reaction 280.
18q22
In the study by
Todd et al. 29, 30,
another locus showed association with T1D: rs763361 in the T lymphocyte costimulation
gene CD226 on chromosome 18q22 (Poverall = 1.38 ![]() 10-8).
CD226 is a glycoprotein expressed on the surface of NK cells, platelets, monocytes
and differentiated Th1 cells. Anti-CD226 treatment has been shown to delay the
onset and reduce the severity of experimental autoimmune encephalomyelitis, a
Th1-mediated autoimmune disease 281.
10-8).
CD226 is a glycoprotein expressed on the surface of NK cells, platelets, monocytes
and differentiated Th1 cells. Anti-CD226 treatment has been shown to delay the
onset and reduce the severity of experimental autoimmune encephalomyelitis, a
Th1-mediated autoimmune disease 281.
IDDM3
The initial evidence for linkage with marker D15S107 on
chromosome 15q26 was initially reported in 250 Caucasian families from the
IDDM4
Several studies reported evidence for the existence of the IDDM4 susceptibility locus. This locus
is tightly linked to the FGF3 marker on chromosome 11q13 284, 286,
288-291. Evidence was found for a decreased transmission (46.4%) to
the affected offspring of a 15 Kb stretch of DNA containing two tightly linked
alleles (D11S1917*03 and H0570polyA*02) 292. In contrast, the D11S1917*03-H0570polyA*02 haplotype
showed increased transmission (56.6%) to unaffected siblings. These results suggest that IDDM4 susceptibility may derive from a
gene very close to the D11S1917 marker. Moreover, similar to that discussed for
IDDM1 and IDDM2, these findings show that analysis of both predisposing and
non-predisposing alleles may be of value when mapping genes for common
polygenic diseases 292. A subsequent study
provided further evidence for linkage with a peak LOD score of 3.4 at the
D11S913 marker 293. Moreover, the
extended transmission disequilibrium test (ETDT) revealed significant
association/linkage with the marker D11S987 (P= 0.0004) within an interval of
approximately 6 cM between D11S4205 and GALN.
Several candidate genes can be found in this chromosomal region. MDU1, encoding a cell-surface cell protein regulating
intracellular calcium, and ZFM1, a nuclear protein, are both expressed in the
pancreas. The RT6 gene lies also in this
region, coding for a T-cell protein that is deficiently expressed in the BB rat
animal model of diabetes 289. The
interleukin-converting enzyme (ICE) and CD3 genes were also proposed as
candidates to explain IDDM4
susceptibility. However, the CD3 gene has been excluded by both association and
linkage analysis 294. A previous report of an association of the CD3 gene with T1D
could have been due to population stratification 295. The gene coding for
the low-density lipoprotein receptor related protein 5 (LRP5) has been mapped
within the boundaries of the IDDM4
locus and proposed as yet another candidate.
However, its functional role in the pathogenesis of T1D remains unclear 296. A large study of markers in the LRP5
region involving 1,106 T1D families provided no further evidence for disease
association at LRP5 or at D11S987. In the same study, the analysis of 1,569
families from
IDDM5
Several but not all studies have
shown linkage with the a046xa9 and ESR markers on chromosome 6q25 166, 286, 288, 291, 301, 302. The
Mn-superoxide dismutase (MnSOD) is a candidate gene for susceptibility at the IDDM5 locus. Polymorphisms affecting the function of MnSOD
could render ß-cells more susceptible to free oxygen radical damage. This
region may contain a susceptibility gene that is common to several autoimmune
diseases 303.
Bohren et al. 304
identified a novel gene, SUMO-4, coding for a Small Ubiquitin-like Modifier 4
protein. The authors also identified a single nucleotide polymorphism involving
a highly conserved methionine with a valine residue (M55V). The SUMO-4 variant
carrying the methionine was associated with diabetes susceptibility in families
from the US and UK
IDDM6
Linkage with the 18q12-q21 region, and in particular with
the Kidd Blood group locus (JK), was suggested almost 20 years ago 310 and linkage to the JK-D18S64 marker was initially confirmed
by the very first genome-wide scan performed in 1994 288. The Transmission Disequilibrium Test (TDT) provided
evidence for increased transmission of allele 4 of marker D18S487 to affected
children in a total of 1,067 families from four different countries. Analysis
using the TDT also provided evidence for genetic heterogeneity, which can often
play as a confounding factor when mapping susceptibility genes in complex
diseases 311. Additional evidence for the existence of the IDDM6 susceptibility locus near D18S487
was provided by another large study of 1,708 families from seven different
countries 312. There is evidence
that this region may predispose to several autoimmune diseases 303. Later studies in
the Finnish population and the 2005 T1DGC genome wide scan did not confirm
evidence for linkage at this locus 45. A candidate
gene has been reported in this region, ZNF236, a gene coding for a Kruppel-like
zinc-finger protein. ZNF236 is ubiquitously expressed in all human tissues
tested. Its expression levels are highest in skeletal muscle and brain,
intermediate in heart, pancreas, and placenta, and lowest in kidney, liver, and
lung. Two alternative spliced forms of the ZNF236 transcript have been found to
be up-regulated in human mesangial cells in response to elevated levels of
glucose, suggesting that ZNF236 may be a candidate gene for diabetic
nephropathy 313.
IDDM7
Linkage on chromosome 2q near the marker D2S326 was
initially reported in
IDDM8
Several groups reported evidence for linkage with markers
D6S264, D6S446 284, 288, and D6S281 286, 301,
302 on
chromosome 6q25-q27 . At present there is no
known candidate gene in the 6q25-q27 region.
Owerbach has defined a linkage disequilibrium map of nearly 1 Mb in the
6q27 region and identified multiple haplotypes associated with IDDM8,
suggesting localization of this putative susceptibility locus to the terminal
200 kb of chromosome 6 323. The IDDM8 locus
may also be subject to parental effects 324 and may confer susceptibility to rheumatoid arthritis as
well 325. Analysis of 831
affected sib pairs in the study of Cox and coworkers implicated IDDM8
only after stratification by HLA genotype 189. Owerbach et al. examined five potential candidate
genes in the IDDM8 region using 36 genetic markers in 478 families and detected
evidence for an association of a CAG/CAA polymorphism in exon 3 of the TATA
box-binding protein gene 326. There is also evidence
that the IDDM8 region contains polymorphisms in the insulin-growth factor II
receptor gene that are associated with increased susceptibility when maternally
transmitted 327.
IDDM9
Initial evidence suggested a susceptibility locus on
chromosome 3q21-q25 in linkage with marker D3S1303 288. IDDM9 appears to
be distinct from a susceptibility locus for Rheumatoid Arthritis reported on
chromosome 3q 328. Laine et al. 329 analyzed
22 microsatellite markers in 121 Finnish type 1 diabetes multiplex families in
the IDDM9 region and detected LOD scores of 3.4 and 2.5 with markers
D3S1589 and D3S3606, respectively. Two additional markers showed association
using the TDT in 384 Finnish type 1 diabetes simplex families. Marker
AFM203wd10 showed association with type 1 diabetes. Interestingly, there was
evidence of interaction with IDDM2. There was no strong evidence of
linkage in the 2005 T1DGC genome scan 45.
IDDM10
Another susceptibility locus may exist on chromosome
10p11-q11 (marker D10S193), and has been termed IDDM10 288. Additional
support for the existence of IDDM10
was provided by the TDT analysis of 1, 159 families with at least one affected
child from the
IDDM11
IDDM11 appears to lie on chromosome 14q24.3-q31 and was linked to
the microsatellite D14S67 using both maximum likelihood methods and affected
sib pair methods. This represents the strongest evidence for linkage to any
locus outside the HLA region. Similar to IDDM3,
the strongest linkage (with the D14S67 marker) was obtained in a subset of
families lacking increased HLA sharing among the affected offspring, suggesting
that IDDM11 may be an important
susceptibility locus in families lacking strong HLA region predisposition 336. Supporting evidence
for IDDM11 has not been replicated in the 2005 T1DGC scan 45. Two
candidate genes have been mapped to this chromosomal region. The ENSA gene
encodes alpha-endosulfine, an endogenous regulator of ß-cell K(ATP) channels 337. The recombinant
alpha-endosulfine has been shown to inhibit sulfanylurea binding to ß-cell
membranes, to reduce cloned K(ATP) channel currents, and to stimulate insulin
secretion from ß-cells. The SEL-1L gene encodes for a negative regulator of the
NOTCH, LIN-12, and GLP-1 receptors, which are required for differentiation and
maturation of cells as well as cell-to-cell interactions during development 297. SEL-1L is abundantly expressed only in the pancreas, and
appears to be involved in the down-regulation of mammalian Notch signaling,
shown to be critical for the development of the pancreas and ß-cells 298. However, a study of
families from
IDDM13
The IDDM13
susceptibility locus lies in the 2q34 region and is linked to the D2S164 marker
in Caucasian families from
IDDM14
This
denomination has not been assigned to any locus.
IDDM15
Linkage with the microsatellite D6S283 on chromosome 6q21
has been reported in families from
IDDM16
Field and coworkers analyzed immunoglobulin heavy chain
(IGH) region microsatellites in 351 North American and British families and 241
families from
IDDM17
Unlike all of the preceding susceptibility loci, which have
been mostly pinpointed by studying large c
IDDM18
Morahan and coworkers 350 reported linkage dysequilibrium between a single base pair
change in the 3’ UTR of the IL12B gene (5q31.1-q33.1) and type 1 diabetes in
two Australian cohorts . This gene
encodes for the p40 subunit of interleukin-12 (IL-12). IL-12 is a
disulphide-linked heterodimer composed of a heavy chain (p40, 40 kDa) and a
light chain (p35, 35 kDa). The IL12A gene located on chromosome 3 encodes the
light chain. The resulting heterodimer
(p70 or p75) is the biologically active form of IL-12. IL-12p40 has been shown
to stimulate Th-1 differentiation and IL-12 accelerates diabetes development in
NOD mice. Thus, the IL-12B gene appears to be an important candidate gene in
terms of immune function. Unfortunately,
multiple studies of family datasets from the
Other Susceptibility Loci
Linkage has been reported with a few other loci that have
not received an official denomination. The glucokinase gene (GCK) on chromosome
7 was linked to IDDM in 339 affected sib-pair families, but this finding has
not been reproduced in other studies 352. Linkage was also reported for the D1S1617 marker on
chromosome 1q (D1S1617) 353, and yet another locus may lie on chromosome X linked to
markers DX6678 and DXS1068. This locus may influence the male-female bias in
HLA-DR3-positive patients 354. The combined
analysis of multiple data sets showed the most dramatic linkage (LOD=3.83)
after IDDM1 and IDDM2 with a region on chromosome 16q22-q24 in association with
D16S3098. This was the only
“significant” LOD score (outside of IDDM1
and IDDM2) in this study of 767
multiplex families of Cox et al. 189. Evidence for
linkage at this locus has been confirmed in the 2005 T1DGC genome scan. In this study, additional chromosomal regions
with linkage to diabetes were 3p13-p14 (D3S1261), 9q33-q34 (D9S260), 12q14-q12
(D12S375), 16p12-q11.1 (D16S3131, 16q22-q24 (D16S504) and 19p13.3-p13.2 (INSR) 45.
PARENTAL EFFECTS ON INHERITED GENES EXPRESSION
The genetics
of type 1 diabetes is further complicated by the possible existence of parental
effects acting on the transmission and expression of inherited genes. Several
studies have shown that diabetes risk differs in the offspring of diabetic
mothers and fathers, although the results of different studies have been discrepant
355, 356. It is also
controversial whether parent of origin effects influence the transmission of IDDM1 alleles to the diabetic offspring 357-359. Moreover, there is evidence that parental origin effects
may be operative at the IDDM8, IDDM10, and IDDM15 loci 302, 324.
Parental
effects also influence the transmission of the VNTR alleles at the IDDM2 locus, and probably this is the
most studied locus in this regard. The first report of linkage at the IDDM2 locus found evidence, in a small
subset of families that were informative for parental origin, that the excess
allele sharing was exclusively paternal 360. Most of the subsequent studies of
intra-familial association demonstrated a statistically significant difference
only for paternally transmitted alleles 153, 361 362. These
observations may be explained by imprinting, a mechanism that regulates gene
expression by silencing either the maternal or the paternal allele. The silencing effect results from the
epigenetic modification (probably mediated by methylation) of the DNA during
the passage from the male or the female germline. This modification of the DNA marks the
genetic material as maternal or paternal (parental imprint). Of note, the insulin gene is located in a
region of the human genome that is known to be subject to parental imprinting 362. The IGF2 gene, which is adjacent to the
insulin gene on chromosome 11p15, was
the first human gene found to be imprinted and it is expressed exclusively from
the paternal chromosome 363. Several other
genes in the region are expressed from the paternal or maternal chromosomes
only, at least in some tissues or developmental stages 364. INS is expressed from both copies in the pancreas of mice 365, human fetuses
of 7-20 weeks gestation 192 and adult
humans 210, 366. However, monoallelic INS expression was observed in the
pancreas of a 40-week old female fetus 366. INS
is also expressed monoallelically, and specifically from the paternal
chromosome in the mouse yolk sack 209. In addition,
evidence has been presented for the imprinted paternal expression of INS in the human yolk sac 367. Thus, imprinted expression can depend
on the tissue and possibly the developmental stage 368. The effects of imprinting on insulin
expression may influence insulin expression during development and
susceptibility to insulin/growth-related diseases in later life, such as
insulin resistance and type 2 diabetes 367. More importantly, it has been shown that INS can be expressed monoallelically in the thymus 88, 89. In all instances identified, the
silenced allele was the one in cis with
a class III VNTR. Such monoallelic
expression resulting from the silencing of class III VNTR transcripts in the
thymus may prevent the protective effect associated with the class III VNTR and
explain the parent-of-origin effects discussed above. Vafiadis et al. studied in more detail the
class III alleles that were silenced in the thymus 369. They developed
a DNA fingerprinting method for identifying the type of alleles corresponding
to the class III VNTR alleles that were found silenced in two thymus samples
(S1, S2), and then analyzed the parental transmission of these type of class
III alleles in a set of 287 diabetic children.
Twelve of 18 possible transmissions of alleles matching the fingerprint
of the S1 or S2 alleles were transmitted to the diabetic offspring, at a
frequency of 0.67, which is significantly higher than the frequency of 0.38
seen in the remaining 142 class III alleles.
These findings suggest that certain class III alleles may be
predisposing instead of protective, and presumably these alleles are silenced
in the thymus with obvious effects on the development of tolerance to
insulin. Moreover, monoallelic INS expression was reported in the
spleen of an 18 year-old Caucasian male, again preventing the expression of the
INS transcript in cis with the class III VNTR allele 366. Assuming that
monoallelic expression in this subject was mediated by imprinting (parents were
unavailable to determine the parental origin of the silenced allele), this
finding suggests that the imprint status may be maintained beyond development
and perhaps throughout life.
There is also evidence for even more complex mechanisms
regulating INS transcription. Bennett et al. 370 studied more than 1,300 triads (two parents and affected
child) and showed that the most common class I VNTR allele among Caucasians,
termed 814 in arbitrary electrophoresis’ mobility units, has a protective
effect similar to that of class III VNTR alleles. A protective effect of the 814 allele was
independently confirmed in Basque families 238. This protective
effect was apparent only when the 814 allele was inherited from fathers with an
814/class III VNTR genotype. In
contrast, fathers with an 814/class I VNTR genotype transmitted both the 814
and other class I VNTR alleles to their diabetic children at similar
frequency. This unusual transmission
pattern suggests that this allele may behave differently in the offspring
depending on the father
OTHER NON-MENDELIAN REGULATORY MECHANISMS
Besides parent of origin effects and other epigenetic
phenomena, there is also evidence that alternative splicing can affect gene
expression in a tissue specific manner and predispose to certain conditions 373. These include type 1 diabetes, multiple sclerosis, and
other neurological diseases 374. In the case of type
1 diabetes, alternative splicing may affect the probability that one would
mount autoimmune responses to the autoantigen IA-2. IA-2 is a tyrosine-phosphatase-like protein
enriched in the secretory granules of islet and neuroendocrine cells and
consists of a single transmembrane (TM) region (residues 577-600) and extra-
and intra-cellular domains 375, 376. An alternatively spliced variant of the IA-2 transcript has
been discovered through the sequencing of a clone (ICA512.bdc) derived from a
human pancreas library that is routinely used as a source of antigen in a
specific assay for the detection IA-2 autoantibodies 377. This alternatively
spliced transcript lacks exon 13 (Dexon 13), which codes for 73
amino acids (aa 557-629) encompassing the TM and juxta-membrane domains. The evaluation of the IA-2 expression in
islets, thymus and spleen from non-diabetic human tissue donors revealed that
thymus and spleen specimens exclusively express the Dexon 13
transcript and lack expression of the full-length transcript. Both transcripts
are expressed in the pancreas. Another alternatively spliced IA-2 transcript in which
129 bp of exon 14 are spliced out, resulting in the deletion of 43 amino acids
(aa 653-695) in the intracellular domain, was detected in about 50% of the
pancreatic samples studied but essentially in none of the thymus and spleen
specimens. Thus, alternative splicing causes differential IA-2 mRNA and
protein expression in pancreas compared to lymphoid organs. Such differences may affect immune
responsiveness to specific epitopes and help explain why IA-2 and not many
other islet proteins become targets of autoimmunity in IDDM. Tolerance to linear
or conformational epitopes typical of the full-length protein or of the Dexon 13 variant
may not be achieved if these epitopes are expressed in islets but not in
lymphoid organs. The specific lack of
expression of the TM/Juxta-membrane domains (exon 13) in lymphoid organs helps
in explaining why epitopes from these domains are often targeted by autoimmune
responses in IDDM 374. Autoantibodies
against IA-2 epitopes encoded by exons 13 and 14 have been reported in patients
and can precede the appearance of autoantibodies against other intra-cellular
epitopes (epitope spreading) 378-381. There is evidence
that the HLA-DR4 restricted, naturally processed 654-674 epitope (exon 14) is
recognized by autoreactive T-cells 382. Similar
to the parent-of-origin effects affecting insulin gene expression in thymus 88 and
peripheral lymphoid organs 89, 366, differential
IA-2 splicing appears to function as mechanism regulating gene expression
independent of inherited alleles at the insulin and IA-2 loci. Although investigations had excluded linkage
with IA-2 polymorphisms 244, these findings
suggest that expression studies for selected candidate genes in tissues
relevant to the disease process can help dissect the complex genetics of a
multi-factorial disease such as type 1 diabetes.
"ACQUIRED" GENETIC POLYMORPHISMS
Factors other than inherited genes must play a role in
determining progression to overt disease in those individuals carrying
predisposing genes. Environmental factors (viruses, diet) are suspected to be
such determinants (reviewed in ref. 383). The ability to identify genetic risk is aiding the search
for environmental factors. It has been suggested that early introduction of
cereals into infant diets dramatically increases development of anti-islet
autoimmunity of high-risk (HLA/family history) individuals 384, 385. Viruses could trigger specific autoimmune responses through
mechanisms of molecular mimicry or by mediating a direct insult to ß-cells. It
is also an intriguing and yet unproven possibility that novel genes may be
acquired through, or their expression stimulated by, environmental factors
(viruses or diet) after birth. Unlike more common viruses, retroviruses can
integrate in the human genome. Retroviral genes can be either inherited or
acquired after birth, and common viral infections and/or sex hormone changes
associated with puberty may activate quiescent retroviruses. Such acquired
expression may trigger the development of diabetes in genetically predisposed
individuals either via cross-reactivity or immunity against novel viral
antigens previously unknown to the immune system. This could drive immunity against
the tissue that is expressing the novel gene, or to any tissue expressing
molecules with significant cross-reactivity. Thus, environmental factors may
provide or activate genes that could act as "disease genes". This
hypothesis was supported by the finding that a human endogenous retrovirus,
termed IDDMK1, 222, is apparently expressed and released from leukocytes in
patients with type 1 diabetes but not in control individuals 386. Yet it
is unclear whether this or similar retroviruses could be expressed in the
endocrine pancreas. It was also
suggested that IDDMK1, 222 could drive the same T-cell receptor restriction
observed in T-lymphocytes infiltrating the endocrine pancreas of two children
who died at the onset of diabetes 387, and
act as a superantigen. However, the role of
IDDMK1, 222 has been questioned by later studies. In fact, IDDMK1, 222 was
found expressed at similar frequencies in patients and controls in several
studies and no evidence for autoreactivity against this virus has been reported
388, 389. The analysis of polymorphisms in the region of the
endogenous retrovirus HERV-K18 or the DNA flanking it, including the CD48 gene,
provided evidence for association of three variants belonging to a single
haplotype. Genotype analysis suggested a dominantly protective effect of this
haplotype. Further genetic and functional analyses are required to confirm
these findings 390.
GENETICS IN DISEASE PREDICTION
With current knowledge, high-risk individuals can be
identified by genetic analysis in the general population 37. Extremely high-risk individuals can be identified
in families. In particular a number of
large population based studies have been carried out stratifying individuals at
birth by HLA genotype and insulin gene polymorphisms. Children born in
In the DAISY study, siblings of patients with type 1
diabetes who have the highest risk HLA genotype (DR3/4-DQ8) have a risk of
activating anti-islet autoimmunity of approximately 50% versus a risk of
approximately 5% for the general population with the same class II HLA
alleles. This dramatic difference in
risk is at present unexplained and we have termed it the “relative
paradox”. A risk exceeding 50% for
children who at birth are characterized only by high-risk class II HLA alleles
(and if both the highest risk DR-DQ alleles and identical by descent for HLA
haplotypes30)
and relation to a proband with type 1 diabetes suggests that if environmental
factors are of importance they are ubiquitous or “family” based. There may be ubiquitous environmental factors
but being ubiquitous they play a minor role in determining familial aggregation
of type 1 diabetes. The difference
between relatives and the general population with the same class II HLA alleles
could also be explained by additional genetic polymorphisms outside the HLA
complex. Combined analysis of
polymorphisms of the insulin and PTPN22 genes may further refine prediction 396. Among first-degree relatives with the high-risk HLA
genotype that were followed for 3 years, 9 of 43 (28.1%) with the high-risk
-23HphI polymorphism developed anti-islet autoantibodies versus two of 36
(5.6%) relatives with the lower-risk -23HphI genotypes. However, PTPN22
polymorphisms did not show a significant difference in risk by genotype in a
study of 85 relatives. Overall, these results highlight the multiplicative risk
of combined high-risk genotypes at different loci in terms of time to
autoantibody and autoimmune disease development.
In
addition, it is plausible that polymorphisms
linked to the HLA complex or modulating the effects of the primary HLA
determinants may have a greater impact on familial aggregation. This hypothesis stems from the observation
that DR3/4-DQ8 siblings of patients with type 1 diabetes in the Denver DAISY
study are almost always HLA identical to their sibling with diabetes. Namely, they share the complete HLA region by
descent with their affected sibling, and thus all polymorphisms in this region
are inherited together. There is growing
evidence that polymorphisms of genes such as DP397, class I HLA, and other genes within this region can
modulate and contribute to risk and these would in families be shared with
patients. In contrast, DR3 and DR4 haplotypes in the general population may not
always carry the full complement of susceptibility alleles. A major effort to further dissect risk
associated with the HLA region remains therefore crucial.
At present we can predict greatly increased risk of type 1 diabetes and a series of other autoimmune disorders by genetic typing at birth, using primarily information provided by HLA DNA based typing. The importance of this information will primarily be driven by our ability to use that information to prevent morbidity and mortality. For some disorders such as celiac disease, strongly associated with HLA-DR3-DQ2 haplotypes, altering the intake of gliadin is an effective therapy, and timing of gliadin introduction may be an important risk factor given genetic susceptibility. For type 1 diabetes we do not at present have a preventive therapy, but participation in studies such as DAISY decrease morbidity at the time of diagnosis 155. Whereas only 1 child of 30 in the DAISY study (HLA typing at birth followed by anti-islet autoantibody determination and metabolic follow up) required hospitalization at the onset of diabetes, approximately 40% of children presenting with diabetes of the general population (without screening) presented with ketoacidosis and required hospitalization 155. As illustrated in Fig. 7.14, many of the children from the general population had glucose greater than 1,000 mg% at diagnosis, and there is an important risk of death from cerebral edema when diagnosis of diabetes is delayed. Of note, even children from the general population with a relative with type 1 diabetes presented with severe metabolic abnormalities. This prevention of onset morbidity will need to be balanced against increased anxiety in families where a child is identified with increased disease risk. We believe it is likely that as the major efforts for prevention and rational treatment of a series of autoimmune diseases are developed, the balance will weigh toward identification, similar to newborn screening for a series of diseases in developed countries.
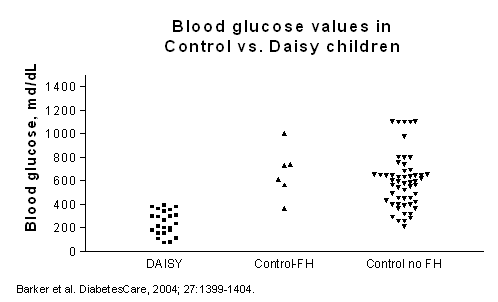
Fig. 7.14
“MONOGENIC” FORMS OF IMMUNE
MEDIATED DIABETES
Dramatic
progress in the understanding of the immunogenetics and pathogenesis of
immune-mediated diabetes has come with the definition of a series of genes in
animal models and man that underlie Mendelian forms of the disease. In particular, two very rare syndromes are
now genetically characterized with plausible mechanistic hypotheses, namely
APS-I (Autoimmune Polyendocrine Syndrome Type 1 (also termed APECED: Autoimmune
Polyendocrinopathy-candidiasis-ectodermal dystrophy; OMIM 240300) 398, 399 and IPEX (immune dysregulation, polyendocrinopathy,
enteropathy, X-linked), also termed the XPID syndrome (X-linked
Polyendocrinopathy, Immune Dysfunction and Diarrhea). The APECED or APS-I syndrome results from
mutations of the AIRE (Autoimmune Regulator) gene. AIRE is a transcription
factor acting as a major (but probably not the only one) determinant of the
development of central thymic tolerance to “peripheral antigens” 400-402, which is mediated by the transcription of genes
coding for peripheral proteins, for example, insulin, in medullary thymic
epithelial cells and dendritic cells 222, 403. The IPEX syndrome results from mutations in
the Foxp3 gene. The Foxp3 gene is essential for the development of regulatory T
lymphocytes 404. Both the
APECED and IPEX syndromes are characterized by the development of immune
mediated diabetes. Neonatal diabetes
develops in patients with the IPEX syndrome while 18% of patients with APS-I
develop diabetes as young children or even adults. Both
syndromes are covered in detail in chapter 8 of this web book. There is
much to learn from these diseases about the pathophysiology of autoimmunity and
key function such as thymic expression of self-molecules (AIRE) and the
generation of regulatory cells (Foxp3) 401. At present there is no or
little indication that polymorphisms at these two loci contribute to common
forms of T1D susceptibility. While one
small case-control study has reported an association of the Foxp3 gene with T1D
in Japanese patients, another study in Sardinian families and a case-control
cohort have not found evidence for linkage or an association with Foxp3 405, 406. Further genetic manipulation of diabetes prone
nonobese diabetic (NOD) mice suggest that Foxp3 does not play a major role in
the spontaneous development of diabetes in a model that closely resembles human
type 1 diabetes 407. However, the administration of Foxp3+CD4+CD25+
regulatory T cells or the administration of T cells transduced with Foxp3 are
reported to antagonize diabetes development in experimental rodent models,
suggesting therapeutic potential even though Foxp3 may be a less specific
marker of regulatory T cells in man 408, 409.
CONCLUDING REMARKS
A large body of evidence indicates that genetic factors influence both susceptibility to and resistance to type 1 diabetes. Several chromosomal regions have been associated with the disease, suggesting that this is a polygenic disorder in most families. Coordinated efforts with large datasets combined with whole genome analyses, are now providing further insight into the genetic factors associated with type 1 diabetes. Mendelian mutations affecting certain genes result in rare monogenic syndromes, the study of which has led to better understanding of the molecular basis of autoimmunity and autoimmune diabetes. These are candidate genes for type 1 diabetes, as polymorphisms may affect their expression and function (albeit less dramatically than in the syndromes) and predispose to type 1 diabetes. Predictably, some of the susceptibility genes for type 1 diabetes are shared with other autoimmune diseases (e.g. PTPN22, CTLA4), while others appear to be disease specific. Based on the information generated so far, almost all of the loci appear to control immune function. It is still possible that some loci may have an effect on selected functions in pancreatic ß-cells, though genetic loci such as TCF7L2 that influences insulin secretion and development of type 2 diabetes is not associated with type 1 diabetes 410. It remains an open question whether no other loci with a major effect exist, similar in risk determination to that of HLA, or whether such loci may exist but be rare variants . In addition we believe it is likely that additional loci with effects potentially larger than those found in the recent Wellcome Trust Whole Genome analysis are present within or linked to the Major Histocompatibility Complex. Defining such loci is complicated by the extensive linkage dysequilibrium in this region that can extend for millions of base pairs. A number of groups are actively pursuing genetic candidates in this region. The ability to predict diabetes with the greatest accuracy based on genetic testing is a critical pre-requisite for the success of primary prevention strategies and, given the dramatic ability to predict risk of type 1A diabetes amongst relatives with the highest risk DR/DQ genotypes, trials for primary prevention with for instance oral insulin (to induce mucosal tolerance: PrePoint) are about to begin based on algorithms identifying extreme genetic risk (determined by having multiple first degree relatives and HLA DR3/4-DQ2/8 or HLA haplotype identity by descent with sibling proband and HLA DR3/4-DQ2/8). A major goal is to define such extreme genetic risk in the general population, and this will almost certainly be dependent upon a fuller understanding of additional polymorphisms contributing to disease that are within or linked to the major histocompatibility complex.
Reference
List
1. Schranz DB, Lernmark A. Immunology in
diabetes: an update. Diabetes Metab Rev 1998;14(1):3-29.
http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=9605628
2. Mordes JP, Bortell R, Blankenhorn EP, Rossini
AA, Greiner DL. Rat models of type 1 diabetes: genetics, environment, and
autoimmunity. ILAR J 2004;45(3):278-291.
3. Eisenbarth GS. Update in type 1 diabetes. J
Clin Endocrinol Metab 2007;92(7):2403-2407.
4. Concannon P, Rich SS, Nepom GT. Genetics of
type 1A diabetes. N Engl J Med 2009;360(16):1646-1654.
5. Barrett JC, Clayton DG, Concannon P et al.
Genome-wide association study and meta-analysis find that over 40 loci affect
risk of type 1 diabetes. Nat Genet 2009.
6. Todd JA. Etiology of type 1 diabetes.
Immunity 2010;32(4):457-467.
7. Erlich H, Valdes AM, Noble J et al. HLA DR-DQ
Haplotypes and Genotypes and Type 1 Diabetes Risk: Analysis of the Type 1
Diabetes Genetics Consortium Families. Diabetes 2008;57:1084-1092.
8. Raj SM, Howson JM, Walker NM et al. No association
of multiple type 2 diabetes loci with type 1 diabetes. diabetol
2009;52(10):2109-2116.
9. Yamamoto N, Fujita Y, Satoh S, Nakanami N,
Sonoda N, Nakano H. Fulminant type 1 diabetes during pregnancy: A case report
and review of the literature. J Obstet Gynaecol Res 2007;33(4):552-556.
10. Murphy R, Ellard S, Hattersley AT. Clinical
implications of a molecular genetic classification of monogenic beta-cell
diabetes. Nat Clin Pract Endocrinol Metab 2008;4(4):200-213.
11. Sagen JV, Raeder H, Hathout E et al.
Permanent neonatal diabetes due to mutations in KCNJ11 encoding Kir6.2: patient
characteristics and initial response to sulfonylurea therapy. Diabetes
2004;53(10):2713-2718.
12. Gianani R, Campbell-Thompson M, Sarkar SA et
al. Dimorphic histopathology of long-standing childhood-onset diabetes.
diabetol 2010.
13. Banerji MA, Chaiken RL, Huey H et al. GAD
antibody negative NIDDM in adult black subjects with diabetic ketoacidosis and
increased frequency of human leukocyte antigen DR3 and DR4. diab
1994;43:741-745.
14. Balasubramanyam A, Nalini R, Hampe CS,
Maldonado M. Syndromes of ketosis-prone diabetes mellitus. Endocr Rev 2008;29(3):292-302.
15. Gilliam LK, Brooks-Worrell BM, Palmer JP,
Greenbaum CJ, Pihoker C. Autoimmunity and clinical course in children with type
1, type 2, and type 1.5 diabetes. J Autoimmun 2005;25(3):244-250.
16. Turner R, Stratton I, Horton V et al. UKPDS 25:
autoantibodies to islet-cell cytoplasm and glutamic acid decarboxylase for
prediction of insulin requirement in type 2 diabetes. UK Prospective Diabetes
Study Group. Lancet 1997;350(9087):1288-1293.
17. Fourlanos S, Dotta F, Greenbaum C et al.
Latent autoimmune diabetes in adults (LADA) should be less latent. diabetol
2005;48:2206-2212.
18. Risch N, Ghosh S, Todd JA. Statistical
evaluation of multiple-locus linkage data in experimental species and its
relevance to human studies: application to nonobese diabetic (NOD) mouse and
human insulin- dependent diabetes mellitus (IDDM). Am J Hum Genet
1993;53:702-714.
19. Todd JA, Farrall M. Panning for gold:
genome-wide scanning for linkage in type I diabetes. Hum Mol Genet
1996;5:1443-1448.
20. Rich SS. Mapping genes in diabetes: genetic
epidemiological perspective. diab 1990;39:1315-1319.
21. Barnett AH, Eff C, Leslie RD, Pyke DA.
Diabetes in identical twins. A study of 200 pairs. diabetol 1981;20(2):87-93.
22. Srikanta S, Ganda OP, Eisenbarth GS, Soeldner
JS. Islet cell antibodies and beta cell function in monozygotic triplets and
twins initially discordant for Type I diabetes mellitus. N Engl J Med
1983;308(6):322-325.
23. Verge CF, Gianani R, Yu L et al. Late
progression to diabetes and evidence for chronic b-cell
autoimmunity in identical twins of patients with type I diabetes. diab
1995;44(10):1176-1179.
24. Redondo MJ, Yu L, Hawa M et al. Heterogeneity
of type I diabetes: analysis of monozygotic twins in Great Britain and the
United States. Diabetologia 2001;44(3):354-362.
25. Olmos P, A'Hearn R, Heaton DA et al. The
significance of the concordance rate of type I (insulin-dependent) diabetes
mellitus in identical twins. diabetol 1988;31(10):747-750.
26. Hearne CM, Ghosh S, Todd JA. Microsatellites
for linkage analysis of genetic traits. TIG 1992;8:288-294.
27. Wellcome Trust Case Control Consortium.
Genome-wide association study of 14,000 cases of seven common diseases and
3,000 shared controls. Nature 2007;447(7145):661-678.
28. Lernmark ADLEGOJPMARPaSR. Human cell lines
from families available for diabetes research. diabetol 1991;34-61.
29. Todd JA, Walker NM, Cooper JD et al. Robust
associations of four new chromosome regions from genome-wide analyses of type 1
diabetes. Nat Genet 2007;39(7):857-864.
30. Aly TA, Ide A, Jahromi MM et al. Extreme
Genetic Risk for Type 1A Diabetes. Proc Natl Acad Sci USA
2006;103(38):14074-14079.
31. Redondo MJ, Rewers M, Yu L et al. Genetic
determination of islet cell autoimmunity in monozygotic twin, dizygotic twin,
and non-twin siblings of patients with type 1 diabetes: prospective twin study.
BMJ 1999;318:698-702.
32. Gale EA, Bingley PJ, Eisenbarth GS, Redondo
MJ, Kyvik KO, Petersen JS. Reanalysis of twin studies suggests that diabetes is
mainly genetic. BMJ 2001;323(7319):997A.
33. Green A, Patterson CC. Trends in the
incidence of childhood-onset diabetes in Europe. diabetol 2001;44(Suppl
3):B3-B8.
34. Bottini N, Musumeci L, Alonso A et al. A
functional variant of lymphoid tyrosine phosphatase is associated with type I
diabetes. Nat Genet 2004;36(4):337-338.
35. Begovich AB, Carlton VE, Honigberg LA et al.
A Missense Single-Nucleotide Polymorphism in a Gene Encoding a Protein Tyrosine
Phosphatase (PTPN22) Is Associated with Rheumatoid Arthritis. Am J Hum Genet
2004;75(2):330-337.
36. Kyogoku C, Langefeld CD, Ortmann WA et al.
Genetic Association of the R620W Polymorphism of Protein Tyrosine Phosphatase
PTPN22 with Human SLE. Am J Hum Genet 2004;75(3):504-507.
37. Lambert AP, Gillespie KM, Thomson G et al.
Absolute Risk of Childhood-Onset Type 1 Diabetes Defined by Human Leukocyte
Antigen Class II Genotype: A Population-Based Study in the United Kingdom. J
Clin Endocrinol Metab 2004;89(8):4037-4043.
38. Baschal EE, Aly TA, Babu SR et al.
HLA-DPB1*0402 Protects Against Type 1A Diabetic Autoimmunity in the Highest
Risk DR3-DQB1*0201/DR4-DQB1*0302 DAISY Population. diab 2007;56(9):2405-2409.
39. Valdes AM, Thomson G, Graham J et al.
D6S265*15 marks a DRB1*15, DQB1*0602 haplotype associated with attenuated
protection from type 1 diabetes mellitus. diabetol 2005;48(12):2540-2543.
40. Johansson S, Lie BA, Todd JA et al. Evidence
of at least two type 1 diabetes susceptibility genes in the HLA complex
distinct from HLA-DQB1, -DQA1 and -DRB1. Genes Immun 2003;4(1):46-53.
41. de Bakker PI, McVean G, Sabeti PC et al. A
high-resolution HLA and SNP haplotype map for disease association studies in
the extended human MHC. Nat Genet 2006;38(10):1166-1172.
42. Lie BA, Todd JA, Pociot F et al. The Predisposition
to Type 1 Diabetes Linked to the Human Leukocyte Antigen Complex Includes at
Least One Non-Class II Gene. Am J Hum Genet 1999; 64:793-800.
43. Lie BA, Sollid LM, Ascher H et al. A gene
telomeric of the HLA class I region is involved in predisposition to both type
1 diabetes and coeliac disease. Tissue Antigens 1999;54(2):162-168.
44. Onengut-Gumuscu S, Concannon P. The genetics
of type 1 diabetes: lessons learned and future challenges. J Autoimmun 2005;25
Suppl:34-39.
45. Concannon P, Erlich HA, Julier C et al. Type
1 diabetes: evidence for susceptibility loci from four genome-wide linkage
scans in 1,435 multiplex families. diab 2005;54(10):2995-3001.
46. Guo D, Li M, Zhang Y et al. A functional
variant of SUMO4, a new IkappaBalpha modifier, is associated with type 1
diabetes. Nat Genet 2004;36(8):837-841.
47. Steenkiste A, Valdes AM, Feolo M et al. 14th
International HLA and Immunogenetics Workshop: report on the HLA component of
type 1 diabetes. Tissue Antigens 2007;69 Suppl 1:214-225.
48. Johnson AH, Hurley CK, Hartzman RJ, Alper CA,
Yunis EJ. HLA: The major histocompatibility complex of man. In: Henry JB,
editor. Clinical Diagnosis & Management by Laboratory Methods. 18 ed.
Philadelphia: WB Saunders; 1991.
49. Noble JA, Valdes AM, Cook M, Klitz W, Thomson
G, Erlich HA. The role of HLA class II genes in insulin-dependent diabetes
mellitus: Molecular analysis of 180 Caucasian, multiplex families. Am J Hum
Genet 1996;59(5):1134-1148.
50. Cudworth AG, Woodrow JC. Evidence for HL-A-linked
genes in "juvenile" diabetes mellitus. Br Med J 1975;3(5976):133-135.
51. Cudworth AG, Woodrow JC. Genetic
susceptibility in diabetes mellitus: analysis of the HLA association. Br Med J
1976;2(6040):846-848.
52. Deschamps I, Lestradet H, Bonaiti C et al.
HLA genotype studies in juvenile insulin-dependent diabetes. diabetol
1980;19(3):189-193.
53. Rotter JI, Anderson CE, Rubin R, Congleton
JE, Terasaki PI, Rimoin DL. HLA genotypic study of insulin-dependent
diabetes. The excess of DR3/DR4 heterozygotes
allows rejection of the recessive hypothesis. diab 1983;32:169.
54. Wolf E, Spencer KM, Cudworth AG. The genetic
susceptibility to Type 1 (insulin-dependent) diabetes: Analysis of the HLA-DR
association. diabetol 1983;24:224-230.
55. Lie BA, Thorsby E. Several genes in the
extended human MHC contribute to predisposition to autoimmune diseases. Curr
Opin Immunol 2005;17(5):526-531.
56. Robles DT, Eisenbarth GS, Wang T et al.
Millennium award recipient contribution. Identification of children with early
onset and high incidence of anti-islet autoantibodies. Clin Immunol
2002;102(3):217-224.
57. Thomson G, Robinson WP, Kuhner MK et al.
Genetic heterogeneity, modes of inheritance, and risk estimates for a joint
study of Caucasians with insulin-dependent diabetes mellitus. Am J Hum Genet
1988;43(6):799-816.
58. Todd JA, Bell JI, McDevitt HO. HLA-DQB gene
contributes to susceptibility and resistance to insulin-dependent diabetes
mellitus. Nature 1987;329(6140):599-604.
59. Horn GT, Bugawan TL, Long CM, Erlich HA.
Allelic sequence variation of the HLA-DQ loci: relationship to serology and to
insulin-dependent diabetes mellitus susceptibility. Proc Natl Acad Sci USA
1988;85(16):6012-6016.
60. Sheehy MJ, Scharf SJ, Rowe JR et al. A
diabetes-susceptible HLA haplotype is best defined by a combination of HLA-DR
and -DQ alleles. J Clin Invest 1989;83(3):830-835.
61. Morel PA, Dorman JS, Todd JA, McDevitt HO,
Trucco M. Aspartic acid at position 57 of the HLA-DQ beta chain protects
against type I diabetes: a family study. Proc Natl Acad Sci USA
1988;85(21):8111-8115.
62. Khalil I, D'Auriol L, Gobet M et al. A
combination of HLA-DQb Asp57-negative and HLA DQa Arg52 confers susceptibility to insulin-dependent diabetes mellitus. J
Clin Invest 1990;85:1315-1319.
63. Khalil I, Deschamps I, Lepage V, Al-Daccak R,
Degos L, Hors J. Dose effect of cis- and trans-encoded HLA-DQ alpha beta
heterodimers in IDDM susceptibility. diab 1992;41:378-384.
64. Ide A, Babu SR, Robles DT et al.
"Extended" A1, B8, DR3 Haplotype Shows Remarkable Linkage
Disequilibrium but Is Similar to Nonextended Haplotypes in Terms of Diabetes
Risk. diab 2005;54(6):1879-1883.
65. Bellgrau D, Pugliese A. NOD mouse and BB rat:
genetics and immunologic function. In: Eisenbarth GS, Lafferty KJ, editors.
Type I Diabetes: Molecular, Cellular, and Clinical Immunology. 1 ed. New York,
New York: Oxford University Press; 1996:53-75.
66. Kwok WW, Domeier ME, Johnson ML, Nepom GT,
Koelle DM. HLA-DQB1 codon 57 is critical for peptide binding and recognition. J
Exp Med 1996;183(3):1253-1258.
67. Sanjeevi CB, DeWeese C, Landin-Olsson M et
al. Analysis of critical residues of HLA-DQ6 molecules in insulin-dependent
diabetes mellitus. Tissue Antigens 1997;50(1):61-65.
68. Hoover ML, Marta RT. Molecular Modelling of
HLA-DQ suggests a mechanism of resistance in type I diabetes. Scand J Immunol
1997;45:193-202.
69. Awata T, Kuzuya T, Matsuda A et al. High
frequency of aspartic acid at position 57 of HLA-DQ B-chain in Japanese IDDM
patients and nondiabetic subjects. diab 1990;39(2):266-269.
70. Erlich HA, Griffith RL, Bugawan TL, Ziegler
R, Alper C, Eisenbarth GS. Implication of specific DQB1 alleles in genetic
susceptibility and resistance by identification of IDDM siblings with novel
HLA-DQB1 allele and unusual DR2 and DR1 haplotypes. diab 1991;40(4):478-481.
71. Zeliszewski D, Tiercy J, Boitard C et al.
Extensive study of DR b,DQ a, and DQ b gene polymorphism in 23 DR2-positive, insulin-dependent diabetes mellitus
patients. Hum Immunol 1992;33(2):140-147.
72. Pugliese A, Zeller M, Yu L, Solimena M,
Ricordi C. Sequence analysis of the DQB1*0602 allele in rare patients with type
1 diabetes. Diabetes 47 (Suppl 1), A198. 1998.
Ref Type: Abstract
73. Pugliese A, Gianani R, Moromisato R et al.
HLA-DQB1*0602 is associated with dominant protection from diabetes even among
islet cell antibody-positive first-degree relatives of patients with IDDM. diab
1995;44(6):608-613.
74. Wen L, Wong FS, Tang J et al. In vivo
evidence for the contribution of human histocompatibility leukocyte antigen
(HLA)-DQ molecules to the development of diabetes. J Exp Med
2000;191(1):97-104.
75. Carrasco-Marin E, Shimizu J, Kanagawa O,
Unanue ER. The class II MHC I-Ag7 molecules from non-obese diabetic
mice are poor peptide binders. J Immunol 1996;156:450-458.
76. Raju R, Munn SR, David CS. T cell recognition
of human pre-proinsulin peptides depends on the polymorphism at HLA DQ
locus: A study using HLA DQ8 and DQ6
transgenic mice. Hum Immunol 1997;58:21-29.
77. Ettinger RA, Kwok WW. A peptide binding motif
for HLA-DQA1*0102/DQB1*0602, the class II MHC molecule associated with dominant
protection in insulin-dependent diabetes mellitus. J Immunol
1998;160(5):2365-2373.
78. Geluk A, van Meijgaarden KE, Schloot NC,
Drijfhout JW, Ottenhoff TH, Roep BO. HLA-DR binding analysis of peptides from
islet antigens in IDDM. diab 1998;47(10):1594-1601.
79. Nishimoto H, Kikutani H, Yamamura K,
Kishimoto T. Prevention of autoimmune insulitis by expression of I-E molecules
in NOD mice. Nature 1987;328(6129):432-434.
80. Koeleman BP, Lie BA, Undlien DE et al.
Genotype effects and epistasis in type 1 diabetes and HLA-DQ trans dimer
associations with disease. Genes Immun 2004;5(5):381-388.
81. Moustakas AK, Papadopoulos GK. Molecular properties
of HLA-DQ alleles conferring susceptibility to or protection from
insulin-dependent diabetes mellitus: keys to the fate of islet beta-cells. Am J
Med Genet 2002;115(1):37-47.
82. Lee KH, Wucherpfennig KW, Wiley DC. Structure
of a human insulin peptide/HLA-DQ8 complex and susceptibility to type 1
diabetes. Nature Immunology 2001;2:501-507.
83. Suri A, Vidavsky I, van der DK, Kanagawa O,
Gross ML, Unanue ER. In APCs, the autologous peptides selected by the
diabetogenic I-Ag7 molecule are unique and determined by the amino acid changes
in the P9 pocket. J Immunol 2002;168:1235-1243.
84. Suri A, Walters JJ, Gross ML, Unanue ER.
Natural peptides selected by diabetogenic DQ8 and murine I-A(g7) molecules show
common sequence specificity. J Clin Invest 2005;115(8):2268-2276.
85. Pugliese A. Genetic Protection from
insulin-dependent diabetes mellitus. Diabetes Nutrition and Metabolism
1997;10:169-179.
86. Baisch JM, Weeks T, Giles R, Hoover M,
Stastny P, Capra JD. Analysis of HLA-DQ genotypes and susceptibility in
insulin- dependent diabetes mellitus. N Engl J Med 1990;322(26):1836-1841.
87. Ronningen KS, Spurkland A, Iwe T, Vartdal F,
Thorsby E. Distribution of HLA-DRB1, -DQA1 and -DQB1 alleles and DQA1-DQB1
genotypes among Norwegian patients with insulin-dependent diabetes mellitus.
Tissue Antigens 1991;37:105-111.
88. Pugliese A, Zeller M, Fernandez A et al. The
insulin gene is transcribed in the human thymus and transcription levels
correlate with allelic variation at the INS VNTR-IDDM2 susceptibility locus for
type I diabetes. Nat Genet 1997;15(3):293-297.
89. Vafiadis P, Bennett ST, Todd JA et al.
Insulin expression in human thymus is modulated by INS VNTR alleles at the
IDDM2 locus. Nat Genet 1997;15:289-292.
90. Rabinovitch A. An update on cytokines in the
pathogenesis of insulin-dependent diabetes mellitus. Diabetes Metab Rev
1998;14(2):129-151.
91. Pugliese A, Kawasaki E, Zeller M et al.
Sequence analysis of the diabetes-protective human leukocyte antigen-DQB1*0602
allele in unaffected, islet cell antibody-positive first degree relatives and
in rare patients with type 1 diabetes. J Clin Endocrinol Metab
1999;84(5):1722-1728.
92. Penny MA, Jenkins D, Mijovic CH et al.
Susceptibility to IDDM in a Chinese population. Role of HLA class II alleles.
diab 1992;41:914-919.
93. Ikegami H, Kawaguchi Y, Yamato E et al.
Analysis by the polymerase chain reaction of histocompatibility leucocyte
antigen-DR9-linked susceptibility to insulin-dependent diabetes mellitus. J
Clin Endocrinol Metab 1992;75(5):1381-1385.
94. Sanjeevi CB, Zeidler A, Shaw S et al.
Analysis of HLA-DQA1 and -DQB1 genes in Mexican Americans with
insulin-dependent diabetes mellitus. Tissue Antigens 1993;42:72-77.
95. Kockum I, Lernmark A, Dahlquist G et al.
Genetic and immunological findings in patients with newly diagnosed
insulin-dependent diabetes mellitus. The Swedish Childhood Diabetes Study Group
and The Diabetes Incidence in Sweden Study (DISS) Group. Horm Metab Res
1996;28(7):344-347.
96. Harbo HF, Lie BA, Sawcer S et al. Genes in
the HLA class I region may contribute to the HLA class II-associated genetic
susceptibility to multiple sclerosis. Tissue Antigens 2004;63:237-247.
97. Slattery RM, Kjer-Nielsen L, Allison J,
Charlton B, Mandel TE, Miller JFAP. Prevention of diabetes in non-obese
diabetic I-Ak transgenic mice. Nature 1990;345(6277):724-726.
98. Schmidt D, Verdaguer J, Averill N, Santamaria
P. A mechanism for the major histocompatibility complex-linked resistance to
autoimmunity. J Exp Med 1997;186:1059-1075.
99. Gianani R, Verge CF, Moromisato-Gianani RI et
al. Limited loss of tolerance to islet autoantigens in ICA+ first degree
relatives of patients with type I diabetes expressing the HLA DQB1*0602 allele.
J Autoimmun 1996;9(3):423-425.
100. Tuomi T, Björses P, Falorni A et al.
Antibodies to glutamic acid decarboxylase and insulin-dependent diabetes in
patients with autoimmune polyendocrine syndrome type I. J Clin Endocrinol Metab
1996;81:1488-1494.
101. Greenbaum CJ, Cuthbertson D, Eisenbarth GS,
Schatz DA, Zeidler A, Krischer JP. Islet cell antibody positive relatives with
HLA-DQA1*0102, DQB1*0602: Identification by the Diabetes Prevention Trial-1. J
Clin Endocrinol Metab 2000;85(3):1255-1260.
102. Redondo MJ, Kawasaki E, Mulgrew CL et al. DR and
DQ associated protection from type 1 diabetes: comparison of DRB1*1401 and
DQA1*0102-DQB1*0602. J Clin Endocrinol Metab 2000;85(10):3793-3797.
103. Caillat-Zucman S, Djilali-Saiah I, Timsit J
et al. Insulin dependent diabetes mellitus (IDDM) joint report. 12th
International Histocompatibility Workshop Study. In: Charron D, editor. Genetic
diversity of HLA. Functional and medical implications. Paris: EDK;
1997:389-398.
104. Erlich HA, Bugawan TL, Scharf S, Nepom GT,
Tait B, Griffith RL. HLA-DQb sequence polymorphism and genetic
susceptibility to IDDM. diab 1990;39:96-103.
105. Nepom GT, Erlich H. MHC class-II molecules
and autoimmunity. Ann Rev Immunol 1991;9:493-525.
106. Tait BD, Drummond BP, Varney MD, Harrison LC.
HLA-DRB1*0401 is associated with susceptibility to insulin-dependent diabetes
mellitus independently of the DQB1 locus. Eur J Immunogenet 1995;22(4):289-297.
107. Cucca F, Muntoni F, Lampis R et al.
Combinations of specific DRB1, DQA1, DQB1 haplotypes are associated with
insulin-dependent diabetes mellitus in Sardinia. Hum Immunol 1993;37(2):85-94.
108. Yasunaga S, Kimura A, Hamaguchi K, Ronningen
KS, Sasazuki T. Different contribution of HLA-DR and -DQ genes in
susceptibility and resistance to insulin-dependent diabetes mellitus (IDDM).
Tissue Antigens 1996;47(1):37-48.
109. Sanjeevi CB, Hook P, Landin-Olsson M et al.
DR4 subtypes and their molecular properties in a population- based study of
Swedish childhood diabetes. Tissue Antigens 1996;47(4):275-283.
110. Undlien DE, Friede T, Rammensee HG et al.
HLA-encoded genetic predisposition in IDDM: DR4 subtypes may be associated with
different degrees of protection. diab 1997;46(1):143-149.
111. Valdes AM, Noble JA, Genin E, Clerget-Darpoux
F, Erlich HA, Thomson G. Modeling of HLA class II susceptibility to Type I
diabetes reveals an effect associated with DPB1. Genet Epidemiol
2001;21(3):212-223.
112. Tait BD, Harrison LC, Drummond BP, Stewart V,
Varney MD, Honeyman MC. HLA antigens and age at diagnosis of insulin-dependent
diabetes mellitus. Hum Immunol 1995;42:116-122.
113. Cruz TD, Valdes AM, Santiago A et al. DPB1
alleles are associated with type 1 diabetes susceptibility in multiple ethnic
groups. diab 2004;53(8):2158-2163.
114. Erlich HA, Rotter JI, Chang JD et al. Association
of HLA-DPB1*0301 with IDDM in Mexican-Americans. diab 1996;45:610-614.
115. Noble JA, Valdes AM, Thomson G, Erlich HA.
The HLA class II locus DPB1 can influence susceptibility to type 1 diabetes.
diab 2000;49(1):121-125.
116. Nejentsev S, Reijonen H, Adojaan B et al. The
effect of HLA-B allele on the IDDM risk defined by DRB1*04 subtypes and
DQB1*0302. diab 1997;46(11):1888-1892.
117. Caillat-Zucman S, Bertin E, Timsit J, Boitard
C, Assan R, Bach JF. Protection from insulin-dependent diabetes mellitus is
linked to a peptide transporter gene. Eur J Immunol 1993;23:1784-1788.
118. Moghaddam PH, de Knijf P, Roep BO et al.
Genetic structure of IDDM1: Two separate regions in the major
histocompatibility complex contribute to susceptibility or protection. diab
1998;47:263-269.
119. Obayashi H, Nakamura N, Fukui M et al.
Influence of TNF microsatellite polymorphisms (TNFa) on age-at-onset of
insulin-dependent diabetes mellitus. Hum Immunol 1999;60(10):974-978.
120. Faustman D, Xiangping L, Lin H et al.
Abnormal MHC class I presentation linked to autoimmunity. Diabetes 41,
supplement 1, 99A. 1992.
Ref Type: Abstract
121. Gaskins HR, Monaco JJ, Leiter EH. Expression
of intra-MHC transporter (Ham) genes and class I antigens in
diabetes-susceptible NOD mice. Science 1992;256:1826-1828.
122. Yan G, Shi L, Faustman D. Novel splicing of
the human MHC-encoded peptide transporter confers unique properties. J Immunol
1999;162(2):852-859.
123. Fu Y, Yan G, Shi L, Faustman D. Antigen
processing and autoimmunity. Evaluation of mRNA abundance and function of
HLA-linked genes. Ann N Y Acad Sci 1998;842:138-155.
124. Yan G, Shi L, Fu Y et al. Screening of the
TAP1 gene by denaturing gradient gel electrophoresis in insulin-dependent
diabetes mellitus: detection and comparison of new polymorphisms between
patients and controls. Tissue Antigens 1997;50(6):576-585.
125. Robinson WP, Barbosa J, Rich SS, Thomson G.
Homozygous parent affected sib pair method for detecting disease predisposing
variants: application to insulin-dependent diabetes mellitus. Genet Epidemiol
1993;10(5):273-288.
126. Chuang LM, Jou TS, Wu HP, Tai TY, Lin BJ. A
rapid method to study heat shock protein 70-2 gene polymorphism in
insulin-dependent diabetes mellitus. Pancreas 1996;13(3):268-272.
127. Karjalainen J, Salmela P, Ilonen J, Surcel
H-M, Knip M. A comparison of childhood and adult type 1 diabetes mellitus. New
Engl J Med 1989;320:881-886.
128. Caillat-Zucman S, Garchon H-J, Timsit J et
al. Age-dependent HLA genetic heterogeneity of type 1 insulin- dependent
diabetes mellitus. J Clin Invest 1992;90:2242-2250.
129. Valdes AM, Thomson G, Erlich HA, Noble JA.
Association between type 1 diabetes age of onset and HLA among sibling pairs.
diab 1999;48(8):1658-1661.
130. Gyllensten UB, Erlich HA. Ancient roots for
polymorphism at the HLA-DQ alpha locus in primates. Proc Natl Acad Sci USA
1989;86:9986-9990.
131. Erlich HA, Gyllensten UB. The evolution of
allelic diversity at the primate major histocompatibility complex class II loci.
Hum Immunol 1991;30:110-118.
132. Honeyman MC, Harrison LC, Drummond B, Colman
PG, Tait BD. Analysis of families at risk for insulin-dependent diabetes
mellitus reveals that HLA antigens influence progression to clinical disease.
Mol Med 1995;1(5):576-582.
133. Noble JA, Valdes AM, Bugawan TL, Apple RJ,
Thomson G, Erlich HA. The HLA class I A locus affects susceptibility to type 1
diabetes. Hum Immunol 2002;63(8):657-664.
134. Valdes AM, Erlich HA, Noble JA. Human
leukocyte antigen class I B and C loci contribute to Type 1 Diabetes (T1D)
susceptibility and age at T1D onset. Hum Immunol 2005;66(3):301-313.
135. Hammond-Kosack MC, Dobrinski B, Lurz R,
Docherty K, Kilpatrick MW. The human insulin gene linked polymorphic region
exhibits an altered DNA structure. Nucleic Acids Res 1992;20(2):231-236.
136. Koeleman BV, De Groot KN, van der Slik AR,
Roep BO, Giphart MJ. Association between D6S2223 and type 1 diabetes
independent of HLA class II in Dutch famlies. diabetol 2002;45:598-599.
137. Valdes AM, Wapelhorst B, Concannon P, Erlich
HA, Thomson G, Noble JA. Extended DR3-D6S273-HLA-B haplotypes are associated
with increased susceptibility to type 1 diabetes in US Caucasians. Tissue
Antigens 2005;65(1):115-119.
138. Gambelunghe G, Ghaderi M, Cosentino A et al.
Association of MHC Class I chain-related A (MIC-A) gene polymorphism with Type
1 diabetes. diabetol 2000;43(4 (2000)):507-514.
139. Bilbao JR, Martin-Pagola A, Calvo B, Perez
dN, Gepv N, Castano L. Contribution of MIC-A polymorphism to type 1 diabetes
mellitus in Basques. Ann N Y Acad Sci 2002;958:321-324.
140. Gupta M, Nikitina-Zake L, Zarghami M et al.
Association between the transmembrane region polymorphism of MHC class I chain
related gene-A and type 1 diabetes mellitus in Sweden. Hum Immunol
2003;64(5):553-561.
141. Gambelunghe G, Falorni A, Ghaderi M et al.
Microsatellite Polymorphism of the MHC Class I Chain-related (MIC-A and MIC-B)
Genes Marks the Risk for Autoimmune Addison's Disease. J Clin Endocrinol Metab
1999;84:3701-3707.
142. Park YS, Sanjeevi CB, Robles D et al.
Additional association of intra-MHC genes, MICA and D6S273, with Addison's
disease. Tissue Antigens 2002;60(2):155-163.
143. Sanjeevi CB, Kanungo A, Berzina L,
Shtauvere-Brameus A, Ghaderi M, Samal KC. MHC class I chain-related gene a
alleles distinguish malnutrition-modulated diabetes, insulin-dependent
diabetes, and non-insulin-dependent diabetes mellitus patients from eastern
India. Ann N Y Acad Sci 2002;958:341-344.
144. Tica V, Nikitina-Zake L, Donadi E, Sanjeevi
CB. MIC-A genotypes 4/5.1 and 9/9 are positively associated with type 1
diabetes mellitus in Brazilian population. Ann N Y Acad Sci 2003;1005:310-313.
145. Novota P, Kolostova K, Pinterova D et al.
Association of MHC class I chain related gene-A microsatellite polymorphism
with the susceptibility to T1DM and LADA in Czech adult patients. Int J
Immunogenet 2005;32:273-275.
146. Ide A, Babu SR, Robles DT et al. Homozygosity
for premature stop codon of the MHC class I chain-related gene A (MIC-A) is
associated with early activation of islet autoimmunity of DR3/4-DQ2/8 high risk
DAISY relatives. J Clin Immunol 2005;25(4):303-308.
147. Caplen NJ, Patel A, Millward A et al.
Complement C4 and heat shock protein 70 (HSP70) genotypes and type I diabetes
mellitus. Immunogenetics 1990;32(6):427-430.
148. Pugliese A, Awdeh Z, Galluzzo A, Yunis EJ,
Alper CA, Eisenbarth GS. No independent association between HSP70 gene
polymorphism and IDDM. diab 1992;41(7):788-791.
149. Cascino I, D'Alfonso S, Cappello N et al.
Gametic association of HSP70-1 promoter region alleles and their inclusion in
extended HLA haplotypes. Tissue Antigens 1993;42(2):62-66.
150. Pociot F, Ronningen KS, Nerup J. Polymorphic
analysis of the human MHC-linked heat shock protein 70 (HSP70-2) and HSP70-Hom
genes in insulin-dependent diabetes mellitus (IDDM). Scand J Immunol
1993;38(5):491-495.
151. Kawaguchi Y, Ikegami H, Fukuda M, Fujioka Y,
Shima K, Ogihara T. Polymorphism of HSP70 gene is not associated with type 1
(insulin-dependent) diabetes mellitus in Japanese. Diabetes Res Clin Pract
1993;21(2-3):103-107.
152. Mijovic CH, Penny MA, Jenkins D et al. The
insulin gene region and susceptibility to insulin-dependent diabetes mellitus
in four races; new insights from Afro-Caribbean race-specific haplotypes.
Autoimmunity 1997;26(1):11-22.
153. Pugliese A, Awdeh ZL, Alper CA, Jackson RA,
Eisenbarth GS. The paternally inherited insulin gene B allele (1,428 FokI site)
confers protection from insulin-dependent diabetes in families. J Autoimmun
1994;7(5):687-694.
154. Bennett ST, Barnett AH, Bain SC, Todd JA.
Dissecting IDDM-2-VNTR-encoded predisposition to type I diabetes. Autoimmunity
1995;21:16.
155. Barker JM, Goehrig SH, Barriga K et al.
Clinical characteristics of children diagnosed with type 1 diabetes through
intensive screening and follow-up. Diab care 2004;27(6):1399-1404.
156. Pugliese A, Bugawan T, Moromisato R et al.
Two subsets of HLA-DQA1 alleles mark phenotypic variation in levels of insulin
autoantibodies in first degree relatives at risk for insulin-dependent
diabetes. J Clin Invest 1994;93:2447-2452.
157. Vehik K, Hamman RF, Lezotte D et al. Trends
in high-risk HLA susceptibility genes among Colorado youth with type 1
diabetes. Diabetes Care 2008;31(7):1392-1396.
158. Hermann R, Knip M, Veijola R et al. Temporal
changes in the frequencies of HLA genotypes in patients with Type 1
diabetes-indication of an increased environmental pressure? diabetol
2003;46(3):420-425.
159. Gillespie KM, Bain SC, Barnett AH et al. The
rising incidence of childhood type 1 diabetes and reduced contribution of
high-risk HLA haplotypes. Lancet 2004;364(9446):1699-1700.
160. Fourlanos S, Varney MD, Tait BD et al. The
rising incidence of type 1 diabetes is accounted for by cases with lower-risk
human leukocyte antigen genotypes. Diab care 2008;31(8):1546-1549.
161. Harjutsalo V, Sjoberg L, Tuomilehto J. Time
trends in the incidence of type 1 diabetes in Finnish children: a cohort study.
Lancet 2008;371(9626):1777-1782.
162. Raum D, Awdeh Z, Yunis EJ, Alper CA, Gabbay
KH. Extended major histocompatibility complex haplotypes in type I diabetes
mellitus. J Clin Invest 1984;74(2):449-454.
163. Dawkins RL, Christiansen FT, Kay PH et al.
Disease associations with complotypes, supratypes and haplotypes. Immunol Rev
1983;70:1-22.
164. Aly TA, Eller E, Ide A et al. Multi-SNP
Analysis of MHC Region: Remarkable Conservation of HLA-A1-B8-DR3 Haplotype.
Diabetes 2006;55(5):1265-1269.
165. Smith WP, Vu Q, Li SS, Hansen JA, Zhao LP,
Geraghty DE. Toward understanding MHC disease associations: Partial
resequencing of 46 distinct HLA haplotypes. Genomics 2006;87(5):561-571.
166. Perez dN, Bilbao JR, Calvo B, Castano L.
Analysis of chromosome 6q in Basque families with type 1 diabetes. GEPV-N. Basque-Navarre
Endocrinology and Paediatric Group. Autoimmunity 2000;33(1):33-36.
167. Palmer JP, Asplin CM, Clemons P et al.
Insulin antibodies in insulin-dependent diabetics before insulin treatment.
Science 1983;222(4630):1337-1339.
168. Vardi P, Ziegler AG, Matthews JH et al.
Concentration of insulin autoantibodies at onset of type I diabetes. Inverse
log-linear correlation with age. Diab care 1988;11(9):736-739.
169. Carroll GJ, Will RK, Peter JB, Garlepp MJ,
Dawkins RL. Penicillamine induced polymyositis and dermatomyositis. J Rheumatol
1987;14(5):995-1001.
170. Yu L, Robles DT, Abiru N et al. Early
expression of antiinsulin autoantibodies of humans and the NOD mouse: evidence
for early determination of subsequent diabetes. Proc Natl Acad Sci USA
2000;97(4):1701-1706.
171. Tree TIM, Peakman M. Autoreactive T cells in
human type 1 diabetes. Endocrinology and Metabolism Clinics of North America
2004;33(1):113-+.
172. Rudy G, Stone N, Harrison LC et al. Similar
peptides from two b cell autoantigens, proinsulin and glutamic acid
decarboxylase, stimulate T cells of individuals at risk for insulin-dependent
diabetes. Mol Med 1995;1(6):625-633.
173. Durinovic-Bello I, I, Boehm BO, Ziegler AG.
Predominantly Recognized ProInsulin T Helper Cell Epitopes in Individuals With
and Without Islet Cell Autoimmunity. J Autoimmun 2002;18(1):55-66.
174. Narendran P, Williams AJ, Elsegood K, Leech
NJ, Dayan CM. Humoral and cellular immune responses to proinsulin in adults
with newly diagnosed type 1 diabetes. Diabetes-Metabolism Research and Reviews
2003;19(1):52-59.
175. Alleva DG, Crowe PD, Jin L et al. A
disease-associated cellular immune response in type 1 diabetics to an
immunodominant epitope of insulin. J Clin Invest 2001;107(2):173-180.
176. Kent SC, Chen Y, Bregoli L et al. Expanded T
cells from pancreatic lymph nodes of type 1 diabetic subjects recognize an
insulin epitope. Nature 2005;435(7039):224-228.
177. Achenbach P, Koczwara K, Knopff A, Naserke H,
Ziegler AG, Bonifacio E. Mature high-affinity immune responses to (pro)insulin
anticipate the autoimmune cascade that leads to type 1 diabetes. J Clin Invest
2004;114(4):589-597.
178. Toma A, Haddouk S, Briand JP et al.
Recognition of a subregion of human proinsulin by class I-restricted T cells in
type 1 diabetic patients. Proc Natl Acad Sci U S A 2005;102(30):10581-10586.
179. Mannering SI, Harrison LC, Williamson NA et
al. The insulin A-chain epitope recognized by human T cells is
posttranslationally modified. J Exp Med 2005;202(9):1191-1197.
180. Wegmann DR, Norbury-Glaser M, Daniel D.
Insulin-specific T cells are a predominant component of islet infiltrates in
pre-diabetic NOD mice. Eur J Immunol 1994;24(8):1853-1857.
181. Haskins K, Wegmann D. Diabetogenic T-cell
clones. diab 1996;45:1299-1305.
182. Wong FS, Karttunen J, Dumont C et al.
Identification of an MHC class I-restricted autoantigen in type 1 diabetes by
screening an organ-specific cDNA library. Nat Med 1999;5(9):1026-1031.
183. Faideau B, Briand JP, Lotton C et al.
Expression of preproinsulin-2 gene shapes the immune response to preproinsulin
in normal mice. J Immunol 2004;172(1):25-33.
184. Moriyama H, Abiru N, Paronen J et al.
Evidence for a primary islet autoantigen (preproinsulin 1) for insulitis and
diabetes in the nonobese diabetic mouse. Proc Natl Acad Sci U S A
2003;100(18):10376-10381.
185. Nakayama M, Abiru N, Moriyama H et al. Prime
role for an insulin epitope in the development of type 1 diabetes in NOD mice.
Nature 2005;435(7039):220-223.
186. Nakayama M, Beilke JN, Jasinski JM et al.
Priming and effector dependence on insulin B:9-23 peptide in NOD islet
autoimmunity. J Clin Invest 2007;117(7):1835-1843.
187. Du W, Wong FS, Li MO et al. TGF-beta
signaling is required for the function of insulin-reactive T regulatory cells.
J Clin Invest 2006;116(5):1360-1370.
188. Jasinski JM, Yu L, Nakayama M et al.
Transgenic insulin (B:9-23) T-cell receptor mice develop autoimmune diabetes
dependent upon RAG genotype, H-2g7 homozygosity, and insulin 2 gene knockout.
Diabetes 2006;55(7):1978-1984.
189. Cox NJ, Wapelhorst B, Morrison VA et al.
Seven regions of the genome show evidence of linkage to type 1 diabetes in a
consensus analysis of 767 multiplex families. Am J Hum Genet
2001;69(4):820-830.
190. Cox NJ, Bell GI, Xiang KS. Linkage
disequilibrium in the human insulin/insulin-like growth factor II region of
human chromosome II. Am J Hum Genet 1988;43(4):495-501.
191. Langholz B, Tuomilehto-Wolf E, Thomas D,
Pitkaniemi J, Tuomilehto J. Variation in HLA-associated risks of childhood
insulin-dependent diabetes in the Finnish population: I. Allele effects at A,
B, and DR loci. DiMe Study Group. Childhood Diabetes in Finland. Genet
Epidemiol 1995;12(5):441-453.
192. Vafiadis P, Bennett ST, Colle E, Grabs R,
Goodyer CG, Polychronakos C. Imprinted and genotype-specific expression of
genes at the IDDM2 locus in pancreas
and leukocytes. J Autoimmun 1996;9:397-403.
193. Stead JD, Buard J, Todd JA, Jeffreys AJ.
Influence of allele lineage on the role of the insulin minisatellite in
susceptibility to type 1 diabetes. Hum Mol Genet 2000;9(20):2929-2935.
194. Stead JD, Jeffreys AJ. Allele diversity and
germline mutation at the insulin minisatellite. Hum Mol Genet 2000;9:713-723.
195. Bell GI, Selby MJ, Rutter WJ. The highly
polymorphic region near the human insulin gene is composed of simple tandemly
repeating sequences. Nature 1982;295(5844):31-35.
196. Kennedy GC, German MS, Rutter WJ. The
minisatellite in the diabetes susceptibility locus IDDM2 regulates insulin
transcription. Nat Genet 1995;9(3):293-298.
197. Lucassen AM, Screaton GR, Julier C, Elliott
TJ, Lathrop M, Bell JI. Regulation of insulin gene expression by the IDDM
associated, insulin locus haplotype. Hum Mol Genet 1995;4(4):501-506.
198. Jolicoeur C, Hanahan D, Smith KM. T-cell
tolerance toward a transgenic b-cell antigen and
transcription of endogenous pancreatic genes in thymus. Proc Natl Acad Sci USA
1994;91(14):6707-6711.
199. Barratt BJ, Payne F, Lowe CE et al. Remapping
the Insulin Gene/IDDM2 Locus in Type 1 Diabetes. diab 2004;53(7):1884-1889.
200. Owerbach D, Aagaard L. Analysis of a 1963-bp
polymorphic region flanking the human insulin gene. Gene 1984;32(3):475-479.
201. Rotwein P, Yokoyama S, Didier DK, Chirgwin
JM. Genetic analysis of the hypervariable region flanking the human insulin
gene. Am J Hum Genet 1986;39(3):291-299.
202. Hammond-Kosack MC, Docherty K. A consensus
repeat sequence from the human insulin gene linked polymorphic region adopts
multiple quadriplex DNA structures in vitro. FEBS Lett 1992;301(1):79-82.
203. Pugliese A, Brown D, Garza D et al.
Self-Antigen Presenting Cells Expressing Islet Cell Molecules in Human Thymus
and Peripheral Lymphoid Organs: Phenotypic Characterization and Implications
for Immunological Tolerance and Type 1 Diabetes. J Clin Invest
2001;107(5):555-564.
204. Sospedra M, Ferrer-Francesch X, Dominguez O,
Juan M, Foz-Sala M, Pujol-Borrell R. Transcription of a broad range of
self-antigens in human thymus suggests a role for central mechanisms in tolerance
toward peripheral antigens. J Immunol 1998;161(11):5918-5929.
205. Bennett ST, Todd JA. Human type 1 diabetes
and the insulin gene: principles of mapping polygenes. Annu Rev Genet
1996;30:343-370.
206. Metcalfe KA, Hitman GA, Rowe RE et al. Concordance
for type 1 diabetes in identical twins is affected by insulin genotype. Diab
care 2001;24(5):838-842.
207. Walter M, Albert E, Conrad M et al.
IDDM2/insulin VNTR modifies risk conferred by IDDM1/HLA for development of Type
1 diabetes and associated autoimmunity. diabetol 2003;46(5):712-720.
208. Halminen M, Veijola R, Reijonen H, Ilonen J,
Akerblom HK, Knip M. Effect of polymorphism in the insulin gene region on IDDM
susceptibility and insulin secretion. The Childhood Diabetes in Finland (DiMe)
Study Group. Eur J Clin Invest 1996;26(10):847-852.
209. Bennett ST, Lucassen AM, Gough SCL et al.
Susceptibility to human type I diabetes at IDDM2 is determined by tandem repeat
variation at the insulin gene minisatellite locus. Nat Genet 1995;9:284-292.
210. Bennett ST, Wilson AJ, Cucca F et al. IDDM2-VNTR-encoded susceptibility to
type 1 diabetes: dominant protection and parental transmission of alleles of
the insulin gene-linked minisatellite locus. J Autoimmun 1996;9:415-421.
211. Lew A, Rutter WJ, Kennedy GC. Unusual DNA
structure of the diabetes susceptibility locus IDDM2 and its effect on
transcription by the insulin promoter factor Pur-1/MAZ. Proc Natl Acad Sci U S
A 2000;97(23):12508-12512.
212. Owerbach D, Gabbay KH. The search for IDDM
susceptibility genes: the next generation. [Review]. diab 1996;45(5):544-551.
213. Sprent J, Webb SR. Intrathymic and
extrathymic clonal deletion of T cells. Curr Opin Immunol 1995;7(2):196-205.
214. Liblau RS, Tisch R, Shokat K et al.
Intravenous injection of soluble antigen induces thymic and peripheral T-cells
apoptosis. Proc Natl Acad Sci U S A 1996;93(7):3031-3036.
215. Heath VL, Moore NC, Parnell SM, Mason DW.
Intrathymic expression of genes involved in organ specific autoimmune disease.
J Autoimmun 1998;11(4):309-318.
216. Werdelin O, Cordes U, Jensen T. Aberrant
expression of tissue-specific proteins in the thymus: A hypothesis for the
development of central tolerance. Scand J Immunol 1998;47:95-100.
217. Pugliese A. Central and peripheral
autoantigen presentation in immune tolerance. Immunology 2004;111:138-146.
218. Smith KM, Olson DC, Hirose R, Hanahan D.
Pancreatic gene expression in rare cells of thymic medulla: evidence for
functional contribution to T cell tolerance. Int Immunol 1997;9(9):1355-1365.
219. Hanahan D. Peripheral-antigen-expressing
cells in thymic medulla: factors in self- tolerance and autoimmunity. Curr Opin
Immunol 1998;10(6):656-662.
220. Throsby M, Homo-Delarche F, Chevenne D, Goya
R, Dardenne M, Pleau JM. Pancreatic hormone expression in the murine thymus:
localization in dendritic cells and macrophages. Endocrinology
1998;139(5):2399-2406.
221. Debinski J, Schulte A, Kyewski B, Klein L.
Promiscuous gene expression in medullary thymic epithelial cells mirrors the
peripheral self. Nat Immunol 2001;2:1032-1039.
222. Garcia CA, Prabakar KR, Diez J et al.
Dendritic cells in human thymus and periphery display a proinsulin epitope in a
transcription-dependent, capture-independent fashion. J Immunol 2005;175:2111-2122.
223. French MB, Allison J, Cram DS et al.
Transgenic expression of mouse proinsulin II prevents diabetes in nonobese
diabetic mice. diab 1996;46:34-39.
224. Chentoufi AA, Polychronakos C. Insulin
expression levels in the thymus modulate insulin-specific autoreactive T-cell
tolerance: the mechanism by which the IDDM2 locus may predispose to diabetes.
diab 2002;51(5):1383-1390.
225. Krishnamurthy B, Dudek NL, McKenzie MD et al.
Responses against islet antigens in NOD mice are prevented by tolerance to
proinsulin but not IGRP. J Clin Invest 2006;116(12):3258-3265.
226. Christofori G, Naik P, Hanahan D.
Deregulation of both imprinted and expressed alleles of the insulin-like growth
factor 2 gene during b-cell tumorigenesis. Nat Genet 1995;10:196-201.
227. Hill DJ, Hogg J. Growth factor control of
pancreatic B cell hyperplasia. Baillieres Clin Endocrinol Metab
1991;5(4):689-698.
228. Nyman T, Pekonen F. The expression of
insulin-like growth factors and their binding proteins in normal human lymphocytes.
Acta Endocrinol (Copenh) 1993;128(2):168-172.
229. Vafiadis P, Grabs R, Goodyer CG, Colle E,
Polychronakos C. A functional analysis of the role of IGF2 in IDDM2-encoded
susceptibility to type 1 diabetes. diab 1998;47(5):831-836.
230. Geenen V, Achour I, Robert F et al. Evidence
that insulin-like growth factor 2 (IGF2) is the dominant thymic peptide of the
insulin superfamily. Thymus 1993;21(2):115-127.
231. Geenen V, Lefebvre PJ. The intrathymic
expression of insulin-related genes: implications for pathophysiology and
prevention of Type 1 diabetes. Diabetes Metab Rev 1998;14(1):95-103.
232. Geenen V, Martens H, Brilot F, Renard C,
Franchimont D, Kecha O. Thymic neuroendocrine self-antigens. Role in T-cell
development and central T-cell self-tolerance. Ann N Y Acad Sci
2000;917:710-723.
233. Kecha-Kamoun O, Achour I, Martens H et al.
Thymic expression of insulin-related genes in an animal model of autoimmune
type 1 diabetes. Diabetes Metab Res Rev 2001;17(2):146-152.
234. Whalen BJ, Marounek J, Weiser P et al. BB rat
thymocytes cultured in the presence of islets lose their ability to transfer
autoimmune diabetes. diab 2001;50(5):972-979.
235. Bieg S, Hanlon C, Hampe CS, Benjamin D,
Mahoney CP. GAD65 and insulin B chain peptide (9-23) are not primary
autoantigens in the type 1 diabetes syndrome of the BB rat. Autoimmunity
1999;31(1):15-24.
236. Mordes JP, Schirf B, Roipko D et al. Oral
insulin does not prevent insulin-dependent diabetes mellitus in BB rats. Ann N
Y Acad Sci 1996;778:418-421.
237. Vang T, Congia M, Macis MD et al.
Autoimmune-associated lymphoid tyrosine phosphatase is a gain-of-function
variant. Nat Genet 2005;37(12):1317-1319.
238. Urrutia I, Calvo B, Bilbao JR, Castano L.
Anomalous behaviour of the 5' insulin gene polymorphism allele 814: lack of
association with Type I diabetes in Basques. GEPV-N Group. Basque-Navarre
Endocrinology and Paediatrics. diabetol 1998;41(9):1121-1123.
239. Hollick JB, Dorweiler JE, Chandler VL.
Paramutation and related allelic interactions. Trends Genet 1997;13(8):302-308.
240. Marron MP, Raffel LJ, Garchon HJ et al.
Insulin-dependent diabetes mellitus (IDDM) is associated with CTLA4
polymorphisms in multiple ethnic groups. Hum Mol Genet 1997;6(8):1275-1282.
241. Awata T, Kurihara S, Iitaka M et al.
Association of CTLA-4 gene A-G polymorphism (IDDM12 locus) with acute-onset and
insulin-depleted IDDM as well as autoimmune thyroid disease (Graves' disease
and Hashimoto's thyroiditis) in the Japanese population. diab
1998;47(1):128-129.
242. Hayashi H, Kusaka I, Nagasaka S et al.
Association of CTLA-4 polymorphism with positive anti-GAD antibody in Japanese
subjects with type 1 diabetes mellitus. Clin Endocrinol (Oxf)
1999;51(6):793-799.
243. Yanagawa T, Maruyama T, Gomi K et al. Lack of
association between CTLA-4 gene polymorphism and IDDM in Japanese subjects.
Autoimmunity 1999;29(1):53-56.
244. Esposito L, Hill NJ, Pritchard LE et al.
Genetic analysis of chromosome 2 in type 1 diabetes: analysis of putative loci
IDDM7, IDDM12, and IDDM13 and candidate genes NRAMP1 and IA-2 and the
interleukin-1 gene cluster. IMDIAB Group. diab 1998;47(11):1797-1799.
245. Klein L, Klugmann M, Nave KA, Tuohy VK,
Kyewski B. Shaping of the autoreactive T-cell repertoire by a splice variant of
self protein expressed in thymic epithelial cells. Nat Med 2000;6:56-61.
246. Badenhoop K, Donner H, Pani M, Rau H,
Siegmund T, Braun J. Genetic susceptibility to type 1 diabetes: clinical and
molecular heterogeneity of IDDM1 and IDDM12 in a german population. Exp Clin
Endocrinol Diabetes 1999;107 Suppl 3:S89-S92.
247. Marron MP, Zeidler A, Raffel LJ et al.
Genetic and physical mapping of a type 1 diabetes susceptibility gene (IDDM12)
to a 100-kb phagemid artificial chromosome clone containing D2S72-CTLA4-D2S105
on chromosome 2q33. diab 2000;49(3):492-499.
248. Hill NJ, Lyons PA, Armitage N, Todd JA,
Wicker LS, Peterson LB. NOD Idd5 locus controls insulitis and diabetes and
overlaps the orthologous CTLA4/IDDM12 and NRAMP1 loci in humans [In Process
Citation]. diab 2000;49(10):1744-1747.
249. Ueda H, Howson JM, Esposito L et al.
Association of the T-cell regulatory gene CTLA4 with susceptibility to
autoimmune disease. Nature 2003;423(6939):506-511.
250. Eggena MP, Walker LS, Nagabhushanam V, Barron
L, Chodos A, Abbas AK. Cooperative roles of CTLA-4 and regulatory T cells in
tolerance to an islet cell antigen. J Exp Med 2004;199(12):1725-1730.
251. Turpeinen H, Laine AP, Hermann R et al. A
linkage analysis of the CTLA4 gene region in Finnish patients with type 1
diabetes. Eur J Immunogenet 2003;30(4):289-293.
252. Blomhoff A, Lie BA, Myhre AG et al.
Polymorphisms in the cytotoxic T lymphocyte antigen-4 gene region confer
susceptibility to Addison's disease. J Clin Endocrinol Metab
2004;89(7):3474-3476.
253. Howson JM, Dunger DB, Nutland S, Stevens H,
Wicker LS, Todd JA. A type 1 diabetes subgroup with a female bias is
characterised by failure in tolerance to thyroid peroxidase at an early age and
a strong association with the cytotoxic T-lymphocyte-associated antigen-4 gene.
Diabetologia 2007;50(4):741-746.
254. Lowe CE, Cooper JD, Brusko T et al.
Large-scale genetic fine mapping and genotype-phenotype associations implicate
polymorphism in the IL2RA region in type 1 diabetes. Nat Genet
2007;39(9):1074-1082.
255. Smyth DJ, Cooper JD, Bailey R et al. A
genome-wide association study of nonsynonymous SNPs identifies a type 1
diabetes locus in the interferon-induced helicase (IFIH1) region. Nat Genet
2006;38(6):617-619.
256. Zipris D, Lien E, Nair A et al. TLR9-signaling
pathways are involved in Kilham rat virus-induced autoimmune diabetes in the
biobreeding diabetes-resistant rat. J Immunol 2007;178(2):693-701.
257. Devendra D, Jasinski J, Melanitou E et al.
Interferon-{alpha} as a Mediator of Polyinosinic:Polycytidylic Acid-Induced
Type 1 Diabetes. diab 2005;54(9):2549-2556.
258. Junttila TT, Laato M, Vahlberg T et al.
Identification of patients with transitional cell carcinoma of the bladder
overexpressing ErbB2, ErbB3, or specific ErbB4 isoforms: real-time reverse
transcription-PCR analysis in estimation of ErbB receptor status from cancer
patients. Clin Cancer Res 2003;9(14):5346-5357.
259. Holbro T, Beerli RR, Maurer F, Koziczak M,
Barbas CF, III, Hynes NE. The ErbB2/ErbB3 heterodimer functions as an oncogenic
unit: ErbB2 requires ErbB3 to drive breast tumor cell proliferation. Proc Natl
Acad Sci U S A 2003;100(15):8933-8938.
260. Vairaktaris E, Goutzanis L, Vassiliou S et
al. Enhancement of erbB2 and erbB3 expression during oral oncogenesis in diabetic
rats. J Cancer Res Clin Oncol 2007.
261. Arai H, Miyamoto K, Taketani Y et al. A
vitamin D receptor gene polymorphism in the translation initiation codon:
effect on protein activity and relation to bone mineral density in Japanese
women. J Bone Miner Res 1997;12(6):915-921.
262. Pani MA, Knapp M, Donner H et al. Vitamin D
receptor allele combinations influence genetic susceptibility to type 1
diabetes in Germans. diab 2000;49(3):504-507.
263. Guja C, Marshall S, Welsh K et al. The study
of CTLA-4 and vitamin D receptor polymorphisms in the Romanian type 1 diabetes
population. J Cell Mol Med 2002;6(1):75-81.
264. Turpeinen H, Hermann R, Vaara S et al.
Vitamin D receptor polymorphisms: no association with type 1 diabetes in the
Finnish population. Eur J Endocrinol 2003;149(6):591-596.
265. Nejentsev S, Cooper JD, Godfrey L et al.
Analysis of the vitamin D receptor gene sequence variants in type 1 diabetes.
diab 2004;53(10):2709-2712.
266. Guo SW, Magnuson VL, Schiller JJ, Wang X, Wu
Y, Ghosh S. Meta-analysis of vitamin D receptor polymorphisms and type 1
diabetes: a HuGE review of genetic association studies. Am J Epidemiol
2006;164(8):711-724.
267. Lopez ER, Zwermann O, Segni M et al. A
promoter polymorphism of the CYP27B1 gene is associated with Addison's disease,
Hashimoto's thyroiditis, Graves' disease and type 1 diabetes mellitus in
Germans. Eur J Endocrinol 2004;151(2):193-197.
268. Bailey R, Cooper JD, Zeitels L et al.
Association of the vitamin D metabolism gene CYP27B1 with type 1 diabetes.
Diabetes 2007;56(10):2616-21.
269. Pozzilli P, Manfrini S, Crino A et al. Low
levels of 25-hydroxyvitamin D3 and 1,25-dihydroxyvitamin D3 in patients with
newly diagnosed type 1 diabetes. Horm Metab Res 2005;37(11):680-683.
270. Littorin B, Blom P, Scholin A et al. Lower
levels of plasma 25-hydroxyvitamin D among young adults at diagnosis of
autoimmune type 1 diabetes compared with control subjects: results from the
nationwide Diabetes Incidence Study in Sweden (DISS). diabetol 2006;49(12):2847-2852.
271. Vitamin D supplement in early childhood and
risk for Type I (insulin-dependent) diabetes mellitus. The EURODIAB Substudy 2
Study Group. diabetol 1999;42(1):51-54.
272. Hypponen E, Laara E, Reunanen A, Jarvelin MR,
Virtanen SM. Intake of vitamin D and risk of type 1 diabetes: a birth-cohort
study. Lancet 2001;358(9292):1500-1503.
273. Jones G, Strugnell SA, DeLuca HF. Current
understanding of the molecular actions of vitamin D. Physiol Rev
1998;78(4):1193-1231.
274. Mathieu C, Waer M, Laureys J, Rutgeerts O,
Bouillon R. Prevention of autoimmune diabetes in NOD mice by 1,25
dihydroxyvitamin D3. diabetol 1994;37(6):552-558.
275. Gregori S, Giarratana N, Smiroldo S,
Uskokovic M, Adorini L. A 1alpha,25-dihydroxyvitamin D(3) analog enhances regulatory
T-cells and arrests autoimmune diabetes in NOD mice. diab 2002;51(5):1367-1374.
276. Fitau J, Boulday G, Coulon F, Quillard T,
Charreau B. The adaptor molecule Lnk negatively regulates tumor necrosis
factor-alpha-dependent VCAM-1 expression in endothelial cells through
inhibition of the ERK1 and -2 pathways. J Biol Chem 2006;281(29):20148-20159.
277. Mashima R, Saeki K, Aki D et al. FLN29, a
novel interferon- and LPS-inducible gene acting as a negative regulator of
toll-like receptor signaling. J Biol Chem 2005;280(50):41289-41297.
278. Mustelin T, Vang T, Bottini N. Protein
tyrosine phosphatases and the immune response. Nat Rev Immunol 2005;5(1):43-57.
279. Mathew PA, Chuang SS, Vaidya SV, Kumaresan
PR, Boles KS, Pham HT. The LLT1 receptor induces IFN-gamma production by human
natural killer cells. Mol Immunol 2004;40(16):1157-1163.
280. Ellery JM, Nicholls PJ. Alternate signalling
pathways from the interleukin-2 receptor. Cytokine Growth Factor Rev
2002;13(1):27-40.
281. Dardalhon V, Schubart AS, Reddy J et al.
CD226 is specifically expressed on the surface of Th1 cells and regulates their
expansion and effector functions. J Immunol 2005;175(3):1558-1565.
282. Dahlman I, Eaves IA, Kosoy R et al.
Parameters for reliable results in genetic association studies in common
disease. Nat Genet 2002;30(2):149-150.
283. Field LL, Tobias R, Magnus T. A locus on
chromosome 15q26 (IDDM3) produces susceptibility to insulin-dependent diabetes
mellitus. Nat Genet 1994;8(2):189-194.
284. Luo D-F, Bui MM, Muir A, Maclaren NK, Thomson
G, She J-X. Affected-sib-pair mapping of a novel susceptibility gene to
insulin-dependent diabetes mellitus (IDDM8) on chromosome 6q25-q27. Am J Hum
Genet 1995;57:911-919.
285. Zamani M, Pociot F, Raeymaekers P, Nerup J,
Cassiman J-J. Linkage of type I diabetes to 15q26 (IDDM3) in the Danish
population. Hum Genet 1996;98:491-496.
286. Luo D-F, Buzzetti R, Rotter JI et al.
Confirmation of three susceptibility genes to insulin-dependent diabetes
mellitus: IDDM4, IDDM5, and IDDM8. Hum Mol Genet 1996;5(5):693-698.
287. Zhoucun A, Zhang S, Xiao C. Preliminary
studies on associations of IDDM3, IDDM4, IDDM5 and IDDM8 with IDDM in Chengdu
population. Chin Med Sci J 2001;16(2):120-122.
288. Davies JL, Kawaguchi S, Bennett ST, Copeman
JB, Cordell HJ, Pritchard P. A genome-wide search for human type 1 diabetes
susceptibility genes. Nature 1994;371:130-136.
289. Hasimoto L, Habita C, Beressi JP et al.
Genetic Mapping of a Susceptibility locus for Insulin-dependent Diabetes Mellitus
on Chromosome 11q. Nature 1994;371(8):161-164.
290. Cordell HJ, Todd JA, Bennett ST, Kawaguchi Y,
Farrall M. Two-locus maximum lod score analysis of a multifactorial trait:
joint consideration of IDDM2 and IDDM4 with IDDM1 in type I diabetes. Am J Hum Genet 1995;57:920-934.
291. Buhler J, Owerbach D, Schaffer AA, Kimmel M,
Gabbay KH. Linkage analyses in type I diabetes mellitus using CASPAR, a
software and statistical program for conditional analysis of polygenic
diseases. Hum Hered 1997;47(4):211-222.
292. Nakagawa Y, Kawaguchi Y, Twells RCJ et al.
Fine mapping of the diabetes-susceptibility locus, IDDM4, on chromosome 11q13.
Am J Hum Genet 1998;63:547-556.
293. Eckenrode S, Marron MP, Nicholls R et al.
Fine-mapping of the type 1 diabetes locus (IDDM4) on chromosome 11q and
evaluation of two candidate genes (FADD and GALN) by affected sibpair and
linkage-disequilibrium analyses. Hum Genet 2000;106(1):14-18.
294. Pritchard LE, Kawaguchi Y, Reed PW et al.
Analysis of the CD3 gene region and type I diabetes: application of
fluorescence-based technology to linkage disequilibrium mapping. Hum Mol Genet
1995;4(2):197-202.
295. Wong S, Moore S, Orisio S, Millward A,
Demaine AG. Susceptibility to type I diabetes in women is associated with the
CD3 epsilon locus on chromosome 11. Clin Exp Immunol 1991;83:69-73.
296. Irwin ML, Ainsworth BE, Stolarczyk LM,
Heyward VH. Predictive accuracy of skinfold equations for estimating body
density of African-American women. Med Sci Sports Exerc 1998;30(11):1654-1658.
297. Harada Y, Ozaki K, Suzuki M et al. Complete
cDNA sequence and genomic organization of a human pancreas-specific gene
homologous to Caenorhabditis elegans sel-1. J Hum Genet 1999;44(5):330-336.
298. Apelqvist A, Li H, Sommer L et al. Notch signalling
controls pancreatic cell differentiation. Nature 1999;400(6747):877-881.
299. Chervonsky AV, Wang Y, Wong FS et al. The
role of Fas in autoimmune diabetes. Cell 1997;89:17-24.
300. Stassi G, De Maria R, Trucco G et al. Nitric
oxide primes pancreatic beta cells for Fas-mediated destruction in
insulin-dependent diabetes mellitus. J Exp Med 1997;186(8):1193-1200.
301. Davies JL, Cucca F, Goy JV et al. Saturation
multipoint linkage mapping of chromosome 6q in type I diabetes. Hum Mol Genet
1996;5(7):1071-1074.
302. Delepine M, Pociot F, Habita C et al.
Evidence of a non-MHC susceptiblity locus in type I diabetes linked to HLA on
chromosome 6. Am J Hum Genet 1997;60:174-187.
303. Vaidya B, Imrie H, Perros P et al. Evidence
for a new Graves disease susceptibility locus at chromosome 18q21. Am J Hum
Genet 2000;66(5):1710-1714.
304. Bohren KM, Nadkarni V, Song JH, Gabbay KH,
Owerbach D. A M55V polymorphism in a novel SUMO gene (SUMO-4) differentially
activates heat shock transcription factors and is associated with
susceptibility to type 1 diabetes mellitus. J Biol Chem 2004;279:27233-27238.
305. Park Y, Park S, Kang J, Yang S, Kim D.
Assessing the validity of the association between the SUMO4 M55V variant and
risk of type 1 diabetes. Nat Genet 2005;37:112-113.
306. Qu H, Bharaj B, Liu XQ et al. Assessing the
validity of the association between the SUM04 M55V variant and risk of type 1
diabetes. Nat Genet 2005;37(2):111-112.
307. Smyth DJ, Howson JM, Lowe CE et al. Assessing
the validity of the association between the SUMO4 M55V variant and risk of type
1 diabetes. Nat Genet 2005;37(2):110-111.
308. Kosoy R, Concannon P. Functional variants in
SUMO4, TAB2, and NFkappaB and the risk of type 1 diabetes. Genes Immun
2005;6(3):231-235.
309. Noso S, Ikegami H, Fujisawa T et al. Genetic
Heterogeneity in Association of the SUMO4 M55V Variant With Susceptibility to
Type 1 Diabetes. diab 2005;54:3582-3586.
310. Hodge SE, Anderson CE, Neiswanger K et al.
Close genetic linkage between diabetes mellitus and kidd blood group. Lancet
1981;2(8252):893-895.
311. Merriman T, Twells R, Merriman M et al.
Evidence by allelic association-dependent methods for a type 1 diabetes
polygene (IDDM6) on chromosome 18q21. Hum Mol Genet 1997;6:1003-1010.
312. Merriman TR, Eaves IA, Twells RC et al.
Transmission of haplotypes of microsatellite markers rather than single marker
alleles in the mapping of a putative type 1 diabetes susceptibility gene
(IDDM6). Hum Mol Genet 1998;7(3):517-524.
313. Holmes DI, Wahab NA, Mason RM. Cloning and
characterization of ZNF236, a glucose-regulated Kruppel-like zinc-finger gene
mapping to human chromosome 18q22-q23. Genomics 1999;60(1):105-109.
314. Copeman JB, Cucca F, Hearne CM et al. Linkage
disequilibrium mapping of a type 1 diabetes susceptibility gene (IDDM7) to
chromosome 2q31-q33. Nat Genet 1995;9(1):80-85.
315. Luo D-F, Maclaren NK, Huang H-S, Muir A, She
J-X. Intrafamilial and case-control association analysis of D2S152 in
insulin-dependent diabetes. Autoimmunity 1995;21:143-147.
316. Kristiansen OP, Pociot F, Bennett EP et al.
IDDM7 links to insulin-dependent diabetes mellitus in Danish multiplex families
but linkage is not explained by novel polymorphisms in the candidate gene
GALNT3. The Danish Study Group of Diabetes in Childhood and The Danish IDDM
Epidemiology and Genetics Group. Hum Mutat 2000;15(3):295-296.
317. Owerbach D, Gabbay KH. The HOXD8 locus (2q31)
is linked to type I diabetes: interaction with chromosome 6 and 11 disease
susceptibility genes. diab 1995;44:132-136.
318. Iwata I, Nagafuchi S, Nakashima H et al.
Association of polymorphism in the NeuroD/BETA2 gene with type 1 diabetes in
the Japanese. diab 1999;48(2):416-419.
319. Hansen L, Jensen JN, Urioste S et al.
NeuroD/BETA2 gene variability and diabetes: no associations to late-onset type
2 diabetes but an A45 allele may represent a susceptibility marker for type 1
diabetes among Danes. Danish Study Group of Diabetes in Childhood, and the
Danish IDDM Epidemiology and Genetics Group. diab 2000;49(5):876-878.
320. Dupont S, Dina C, Hani EH, Froguel P. Absence
of replication in the French population of the association between beta
2/NEUROD-A45T polymorphism and type 1 diabetes. Diabetes Metab
1999;25(6):516-517.
321. Malecki MT, Klupa T, Moczulski DK, Rogus JJ.
The Ala45Thr polymorphism of BETA2/NeuroD1 gene and susceptibility to type 1
diabetes mellitus in caucasians. Exp Clin Endocrinol Diabetes
2003;111(5):251-254.
322. Bennett EP, Hassan H, Mandel U et al. Cloning
and characterization of a close homologue of human
UDP-N-acetyl-alpha-D-galactosamine:Polypeptide
N-acetylgalactosaminyltransferase-T3, designated GalNAc-T6. Evidence for
genetic but not functional redundancy. J Biol Chem 1999;274(36):25362-25370.
323. Owerbach D. Physical and genetic mapping of
IDDM8 on chromosome 6q27. diab 2000;49(3):508-512.
324. Paterson AD, Naimark DM, Petronis A. The
analysis of parental origin of alleles may detect susceptibility loci for
complex disorders. Hum Hered 1999;49(4):197-204.
325. Myerscough A, John S, Barrett JH, Ollier WE,
Worthington J. Linkage of rheumatoid arthritis to insulin-dependent diabetes
mellitus loci: evidence supporting a hypothesis for the existence of common
autoimmune susceptibility loci. Arthritis Rheum 2000;43(12):2771-2775.
326. Owerbach D, Pina L, Gabbay KH. Association of
a CAG/CAA repeat sequence in the TBP gene with type 1 diabetes. Biochem Biophys
Res Commun 2004;323:865-869.
327. McCann JA, Xu YQ, Frechette R, Guazzarotti L,
Polychronakos C. The insulin-like growth factor-II receptor gene is associated
with type 1 diabetes: evidence of a maternal effect. J Clin Endocrinol Metab
2004;89:5700-5706.
328. Paterson AD, Rahman P, Petronis A. IDDM9 and
a locus for rheumatoid arthritis on chromosome 3q appear to be distinct. Hum
Immunol 1999;60(9):883-885.
329. Laine AP, Turpeinen H, Veijola R et al.
Evidence for linkage to an association with type 1 diabetes at the 3q21 region
in the Finnish population. Genes Immun 2006;7:69-72.
330. Reed P, Cucca F, Jenkins S et al. Evidence
for a type 1 diabetes susceptibility locus (IDDM10) on human chromosome
10p11-q11. Hum Mol Genet 1997;6:1011-1016.
331. Chistiakov DA, Seryogin Y, Savost'anov KV et
al. Evidence for a Type 1 Diabetes Susceptibility Locus (IDDM10) on Chromosome
10p11-q11 in a Russian Population. Scand J Immunol 2004;60(3):316-323.
332. Johnson GC, Koeleman BP, Todd JA. Limitations
of stratifying sib-pair data in common disease linkage studies: an example
using chromosome 10p14-10q11 in type 1 diabetes. Am J Med Genet
2002;113(2):158-166.
333. Paterson AD, Petronis A. Age of
diagnosis-based linkage analysis in type 1 diabetes. Eur J Hum Genet
2000;8(2):145-148.
334. Ide A, Kawasaki E, Abiru N et al.
Stromal-cell derived factor-1 chemokine gene variant is associated with type 1
diabetes age at onset in Japanese population. Hum Immunol 2003;64:973-978.
335. Nejentsev S, Smink LJ, Smyth D et al.
Sequencing and association analysis of the type 1 diabetes-linked region on
chromosome 10p12-q11. BMC Genet 2007;8:24.:24.
336. Field LL, Tobias R, Thomson G, Plon S.
Susceptibility to insulin-dependent diabetes mellitus maps to a locus (IDDM11) on human chromosome
14q24.3-q31. Genomics 1996;33:1-8.
337. Heron L, Virsolvy A, Apiou F, Le Cam A,
Bataille D. Isolation, characterization, and chromosomal localization of the
human ENSA gene that encodes alpha-endosulfine, a regulator of beta-cell K(ATP)
channels. diab 1999;48(9):1873-1876.
338. Pociot F, Larsen ZM, Zavattari P et al. No
evidence for SEL1L as a candidate gene for IDDM11-conferred susceptibility.
Diabetes Metab Res Rev 2001;17(4):292-295.
339. Hawkes CJ, Schloot NC, Marks JB et al. T-Cell
Lines Reactive to an Immunodominant Epitope of the Tyrosine Phosphatase-Like
Autoantigen IA-2 in Type 1 Diabetes. diab 2000;49:356-366.
340. Paterson AD, Petronis A. Sex of affected
sibpairs and genetic linkage to type 1 diabetes. Am J Med Genet
1999;84(1):15-19.
341. Fu J, Ikegami H, Kawaguchi Y et al.
Association of distal chromosome 2q with IDDM in Japanese subjects. diabetol
1998;41:228-232.
342. Dotta F, Dionisi S, Viglietta V et al. T-cell
mediated autoimmunity to the insulinoma-associated protein 2 islet tyrosine
phosphatase in type 1 diabetes mellitus. Eur J Endocrinol 1999;141(3):272-278.
343. Owerbach D, Naya FJ, Tsai MJ, Allander SV,
Powell DR, Gabbay KH. Analysis of candidate genes for susceptibility to type I
diabetes: a case-control and family-association study of genes on chromosome
2q31-35. diab 1997;46(6):1069-1074.
344. Nerup J, Pociot F. A genomewide scan for type
1-diabetes susceptibility in Scandinavian families: identification of new loci
with evidence of interactions. Am J Hum Genet 2001;69(6):1301-1313.
345. Temple IK, James RS, Crolla JA et al. An
imprinted gene(s) for diabetes? Nat Genet 1995;9(2):110-112.
346. Haig D. Is human insulin imprinted? Nat Genet
1994;7(1):10.
347. Field LL, Larsen Z, Pociot F, Nerup J, Tobias
R, Bonnevie-Nielsen V. Evidence for a locus (IDDM16) in the immunoglobulin
heavy chain region on chromosome 14q32.3 producing susceptibility to type 1
diabetes. Genes Immun 2002;3(6):338-344.
348. Verge CF, Vardi P, Babu S et al. Evidence for
oligogenic inheritance of type 1A diabetes in a large Bedouin Arab family. J
Clin Invest 1998;102(8):1569-1575.
349. Eller E, Vardi P, Daly MJ et al. IDDM17:
polymorphisms in the AMACO gene are associated with dominant protection against
type 1A diabetes in a Bedouin Arab family. Ann N Y Acad Sci
2004;1037:145-9.:145-149.
350. Lin DY, Feuer EJ, Etzioni R, Wax Y.
Estimating medical costs from incomplete follow-up data. Biometrics
1997;53:419-434.
351. Murphy VJ, Harrison LC, Rudert WA et al.
Retroviral superantigens and type 1 diabetes mellitus. Cell 1998;95(1):9-11.
352. Muir A, Ruan QG, Marron MP, She JX. The
IDDMK(1,2)22 retrovirus is not detectable in either mRNA or genomic DNA from
patients with type 1 diabetes. diab 1999;48(1):219-222.
353. Badenhoop K, Donner H, Neumann J et al. IDDM
patients neither show humoral reactivities against endogenous retroviral
envelope protein nor do they differ in retroviral mRNA expression from healthy
relatives or normal individuals. diab 1999;48(1):215-218.
354. Cucca F, Goy JV, Kawaguchi Y et al. A
male-female bias in type 1 diabetes and linkage to chromosome Xp in MHC
HLA-DR3-positive patients. Nat Genet 1998;19:301-302.
355. Warram JH, Martin BC, Krolewski AS. Risk of
IDDM in children of diabetic mothers decreases with increasing maternal age at
pregnancy. diab 1991;40:1679-1684.
356. Bleich D, Polak M, Eisenbarth GS, Jackson RA.
Decreased risk of type I diabetes in offspring of mothers who acquire diabetes
during adrenarchy. diab 1993;42:1433-1439.
357. Vadheim CM, Rotter JI, Maclaren NK, Riley WJ,
Anderson CE. Preferential transmission of diabetic alleles within the HLA gene
complex. N Engl J Med 1986;315(21):1314-1318.
358. Deschamps I, Hors J, Clerget-Darpoux F et al.
Excess of maternal HLA-DR3 antigens in HLA DR3,4 positive type 1
(insulin-dependent) diabetic patients. diabetol 1990;33(7):425-430.
359. Bain SC, Rowe BR, Barnett AH, Todd JA.
Parental origin of diabetes-associated HLA types in sibling pairs with type I
diabetes. diab 1994;43:1462-1468.
360. Julier C, Hyer RN, Davies RN et al.
Insulin-IGF2 region on chromosome 11p encodes a gene implicated in HLA-DR4-dependent
diabetes susceptibility. Nature 1991;354:155-159.
361. Bui MM, Luo D-F, She JY et al. Paternally
transmitted IDDM2 influences diabetes
susceptibility despite biallelic expression of the insulin gene in human
pancreas. J Autoimmun 1996;9:97-103.
362. Hoffman AR, Vu TH. Genomic imprinting.
Scientific Amer 1996;52-61.
363. Giannoukakis N, Deal C, Paquette J, Goodyer
CG, Polychronakos C. Parental genomic imprinting of the human IGF2 gene. Nat
Genet 1993;4:98-101.
364. Matsuoka S, Thompson JS, Edwards MC et al.
Imprinting of the gene encoding a human cyclin-dependent kinase inhibitor,p57KIP2,
on chromosome 11p15. Proc Natl Acad Sci USA 1996;93:3026-3030.
365. Giddings SJ, King CD, Harman KW, Flood JF,
Carnaghi LR. Allele specific inactivation of insulin 1 and 2, in the mouse yolk
sac, indicates imprinting. Nat Genet 1994;6:310-313.
366. Miceli D, Zeller D, Brown C, Ricordi C,
Pugliese A. Insulin gene expression and imprinting in pancreas and lymphoid
organs. Diabetes 48 (Suppl. 1), A190. 1999.
Ref Type: Abstract
367. Moore GE, Abu-Amero SN, Bell G et al.
Evidence that insulin is imprinted in the human yolk sac. diab
2001;50(1):199-203.
368. Deltour L, Montagutelli X, Guenet JL, Jami J,
Paldi A. Tissue- and developmental stage-specific imprinting of the mouse
proinsulin gene, Ins2. Dev Biol 1995;168(2):686-688.
369. Vafiadis P, Ounissi-Benkalha H, Palumbo M et
al. Class III alleles of the variable number of tandem repeat insulin
polymorphism associated with silencing of thymic insulin predispose to type 1
diabetes. J Clin Endocrinol Metab 2001;86(8):3705-3710.
370. Bennett ST, Wilson AJ, Esposito L et al.
Insulin VNTR allele-specific effect in type 1 diabetes depends on identity of
untransmitted paternal allele. The
IMDIAB Group. Nat Genet 1997;17:350-352.
371. Duvillie B, Bucchini D, Tang T, Jami J, Paldi
A. Imprinting at the mouse Ins2 locus: evidence for cis- and trans-allelic
interactions. Genomics 1998;47(1):52-57.
372. LaSalle JM, Lalande M. Homologous association
of oppositely imprinted chromosomal domains. Science 1996;272(5262):725-728.
373. Black DL. Protein diversity from alternative
splicing: a challenge for bioinformatics and post-genome biology. Cell
2000;103(3):367-370.
374. Diez J, Park Y, Zeller M et al. Differential
splicing of the IA-2 mRNA in pancreas and lymphoid organs as a permissive
genetic mechanism for autoimmunity against the IA-2 type 1 diabetes
autoantigen. diab 2001;50(4):895-900.
375. Rabin DU, Pleasic SM, Shapiro JA et al. Islet
cell antigen 512 is a diabetes-specific islet autoantigen related to protein
tyrosine phosphatases. J Immunol 1994;152(6):3183-3188.
376. Payton MA, Hawkes CJ, Christie MR.
Relationship of the 37,000- and 40,000-Mr tryptic fragments of islet
antigens in insulin-dependent diabetes to the protein tyrosine phosphatase-like
molecule IA-2 (ICA512). J Clin Invest 1995;96:1506-1511.
377. Park YS, Kawasaki E, Kelemme K et al. Humoral
autoreactivity to an alternative spliced variant of ICA512/IA2 in type 1
diabetes. diabetol 2000;50:895-900.
378. Bearzatto M, Naserke H, Piquer S et al. Two
distinctly HLA-associated contiguous linear epitopes uniquely expressed within
the islet antigen 2 molecule are major autoantibody epitopes of the
diabetes-specific tyrosine phosphatase-like protein autoantigens. Journal of
Immunology 2002;168(8):4202-4208.
379. Naserke HE, Ziegler AG, Lampasona V,
Bonifacio E. Early development and spreading of autoantibodies to epitopes of
IA-2 and their association with progression to type 1 diabetes. J Immunol
1998;161(12):6963-6969.
380. Bonifacio E, Christie MR. Tyrosine
phosphatase-like proteins as autoantigens in insulin-dependent mellitus: the
targets for 37/40K antibodies. Diab Nutr Metab 1996;9:183-187.
381. Lampasona V, Bearzatto M, Genovese S, Bosi E,
Ferrari M, Bonifacio E. Autoantibodies in insulin-dependent diabetes recognize
distinct cytoplasmic domain of the protein tyrosine phosphatase-like IA-2
autoantigen. J Immunol 1996;157:2707-2711.
382. Peakman M, Stevens EJ, Lohmann T et al. Naturally
processed and presented epitopes of the islet cell autoantigen IA-2 eluted from
HLA-DR4 [see comments]. J Clin Invest 1999;104(10):1449-1457.
383. Honeyman M. How robust is the evidence for
viruses in the induction of type 1 diabetes? Curr Opin Immunol 2005;17:616-623.
384. Norris JM, Barriga K, Klingensmith G et al.
Timing of cereal exposure in infancy and risk of islet autoimmunity. The Diabetes Autoimmunity Study in the Young
(DAISY). JAMA 2003;290(13):1713-1720.
385. Ziegler AG, Schmid S, Huber D, Hummel M,
Bonifacio E. Early infant feeding and risk of developing type 1
diabetes-associated autoantibodies. JAMA 2003;290(13):1721-1728.
386. Conrad B, Weissmahr RN, Böni J, Arcari R,
Schüpbach J, Mach B. A human endogenous retroviral superantigen as candidate
autoimmune gene in type 1 diabetes. Cell 1997;90(2):303-313.
387. Conrad B, Weidmann E, Trucco G et al.
Evidence for superantigen involvement in insulin-dependent diabetes mellitus
aetiology. Nature 1994;371(6495):351-355.
388. Knerr I, Repp R, Dotsch J et al. Quantitation
of gene expression by real-time PCR disproves a "retroviral
hypothesis" for childhood-onset diabetes mellitus. Pediatr Res
1999;46(1):57-60.
389. Kim A, Jun HS, Wong L et al. Human endogenous
retrovirus with a high genomic sequence homology with IDDMK(1,2)22 is not
specific for Type I (insulin-dependent) diabetic patients but ubiquitous.
diabetol 1999;42(4):413-418.
390. Marguerat S, Wang WY, Todd JA, Conrad B.
Association of human endogenous retrovirus K-18 polymorphisms with type 1
diabetes. diab 2004;53(3):852-854.
391. Rewers M, Bugawan TL, Norris JM et al.
Newborn screening for HLA markers associated with IDDM: diabetes autoimmunity
study in the young (DAISY). diabetol 1996;39(7):807-812.
392. Jorns A, Kubat B, Tiedge M et al. Pathology
of the pancreas and other organs in the diabetic LEW.1AR1/Ztm- iddm rat, a new
model of spontaneous insulin-dependent diabetes mellitus. Virchows Arch
2004;444(2):183-189.
393. Hummel M, Bonifacio E, Schmid S, Walter M,
Knopff A, Ziegler AG. Brief communication: Early appearance of islet
autoantibodies predicts childhood type 1 diabetes in offspring of diabetic
parents. Annals of Internal Medicine 2004;140(11):882-886.
394. Ferber KM, Keller E, Albert ED, Ziegler AG.
Predictive value of human leukocyte antigen class II typing for the development
of islet autoantibodies and insulin-dependent diabetes postpartum in women with
gestational diabetes. J Clin Endocrinol Metab 1999;84(7):2342-2348.
395. Hermann R, Bartsocas CS, Soltesz G et al.
Genetic screening for individuals at high risk for type 1 diabetes in the
general population using HLA Class II alleles as disease markers. A comparison
between three European populations with variable rates of disease incidence.
Diabetes Metab Res Rev 2004;20(4):322-329.
396. Aly T, Ide A, Humphrey K. et al. Genetic
prediction of autoimmunity: Initial oligogenic prediction of anti-islet
autoimmunity amongst DR3/DR4-DQ8 relatives of patients with type 1A diabetes. J
Autoimmun 2005;25(Suppl):40-45.
397. Siegler M, Amiel S, Lantos J. Scientific and
ethical consequences of disease prediction. diabetol 1992;35(Suppl 2):S60-S68.
398. Halonen M, Eskelin P, Myhre AG et al. AIRE
Mutations and Human Leukocyte Antigen Genotypes as Determinants of the
Autoimmune Polyendocrinopathy-Candidiasis-Ectodermal Dystrophy Phenotype. J
Clin Endocrinol Metab 2002;87(6):2568-2574.
399. Perheentupa J. APS-I/APECED: The clinical
disease and therapy. In: Eisenbarth GS, editor. Autoimmune Polyendocrine
Syndromes. Philadelphia: W.B. Saunders Company; 2002:295-320.
400. Mathis D, Benoist C. Back to central
tolerance. Immunity 2004;20(5):509-516.
401. Anderson M, Venanzi ES, Chen Z, Berzins SP,
Benoist C, Mathis D. The cellular mechanism of Aire control of T cell
tolerance. Immunity 2005;23:227-239.
402. Gotter J, Brors B, Hergenhahn M, Kyewski B.
Medullary epithelial cells of the human thymus express a highly diverse
selection of tissue-specific genes coloalized in chromosomal clusters. J Exp
Med 2004;199:155-166.
403. Derbinski J, Gabler J, Brors B et al.
Promiscuous gene expression in thymic epithelial cells is regulated at multiple
levels. J Exp Med 2005;202:33-45.
404. Fontenot JD, Gavin MA, Rudensky AY. Foxp3
programs the development and function of CD4+CD25+ regulatory T cells. Nat
Immunol 2003;4(4):330-336.
405. Bassuny WM, Ihara K, Sasaki Y et al. A
functional polymorphism in the promoter/enhancer region of the FOXP3/Scurfin
gene associated with type 1 diabetes. Immunogenetics 2003;55(3):149-156.
406. Zavattari P, Deidda E, Pitzalis M et al. No
association between variation of the FOXP3 gene and common type 1 diabetes in
the Sardinian population. diab 2004;53(7):1911-1914.
407. Chen Z, Herman AE, Matos M, Mathis D, Benoist
C. Where CD4+CD25+ T reg cells impinge on autoimmune diabetes. J Exp Med
2005;202(10):1387-1397.
408. Lundsgaard D, Holm TL, Hornum L, Markholst H.
In vivo control of diabetogenic T-cells by regulatory CD4+CD25+ T-cells
expressing Foxp3. diab 2005;54(4):1040-1047.
409. Jaeckel E, von Boehmer H, Manns MP.
Antigen-Specific FoxP3-Transduced T-Cells Can Control Established Type 1
Diabetes. diab 2005;54(2):306-310.
410. Field SF, Howson JM, Smyth DJ, Walker NM,
Dunger DB, Todd JA. Analysis of the type 2 diabetes gene, TCF7L2, in 13,795
type 1 diabetes cases and control subjects. diabetol 2007;50(1):212-213.